







吸附是催化、腐蚀等领域绕不开的话题,差分电荷密度广泛用于研究体系吸附前后电荷密度差异,进而可以用于揭示反应机理。
目录
1 差分电荷密度(Differential Charge Density)
1 差分电荷密度(Differential Charge Density)
概念:一个体系在经过某种操作(如吸附、替代等)后,操作后的电荷密度减去操作前的电荷密度所得到的电荷密度分布差。
- 基于电子结构理论:在量子力学中,通过求解薛定谔方程可得到电子电荷密度,但通常计算的是外层价电子的薛定谔方程,而将内层电子当作芯电子不参与求解。当体系发生如吸附或原子替代等过程时,原子核与芯电子构成的原子实位置不变,此时电荷的分布的变化主要源于价电子分布的改变,这种价电子分布变化所体现出的前后电荷密度差异就是差分电荷密度。
- 用于分析化学过程:差分电荷密度能够直观地展现一个化学过程中的电荷转移情况,例如在吸附过程中,通过计算吸附前后的差分电荷密度,可以清楚地看到吸附分子与吸附表面之间的电荷聚集或减少的区域,从而判断成键状况以及相互作用的强弱。
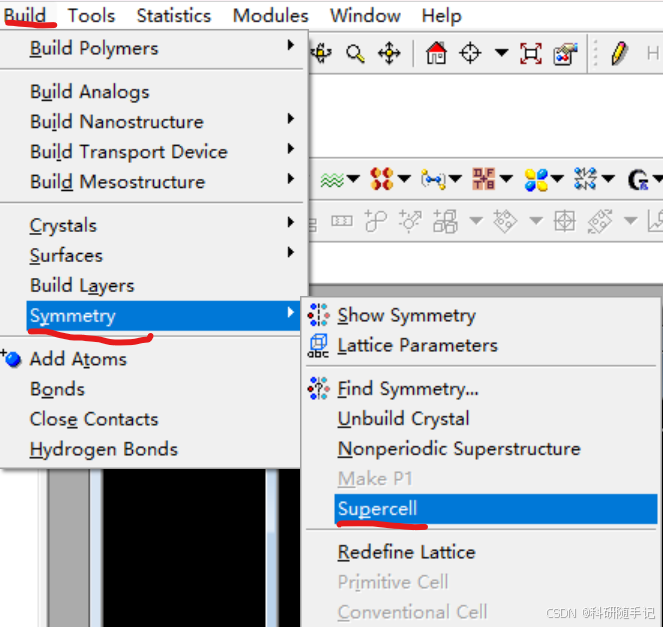
2 计算流程
2.1 搭建CO-Pt的slab结构
2.1.1 构建Pt单胞
打开materials studio→在File中点击Import→选择Structures→选择metals→选择pure-metals→找到并打开Pt.msi
 |
|
|
|
|
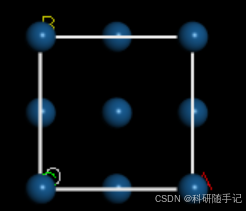
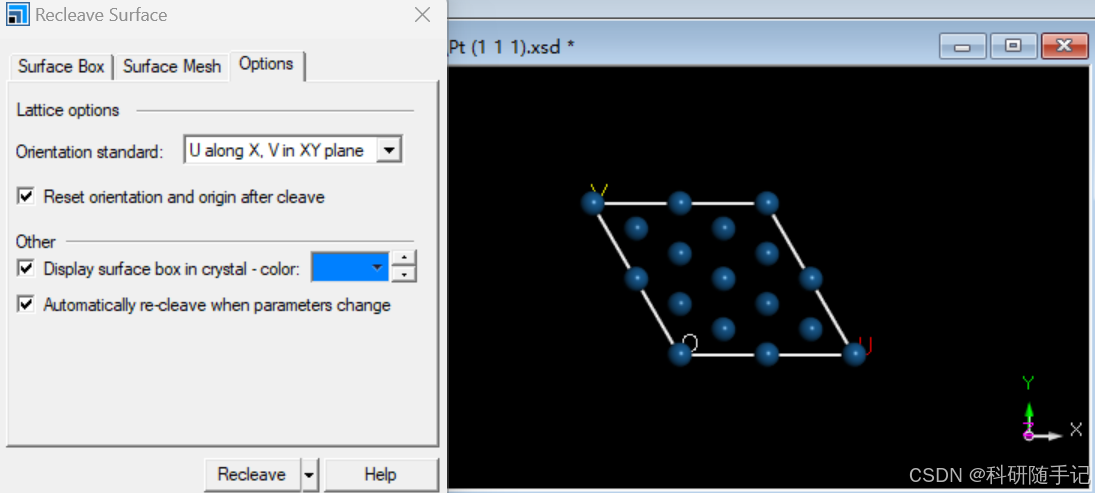
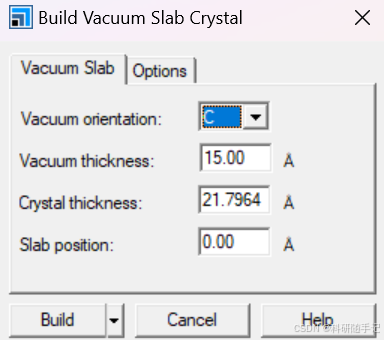
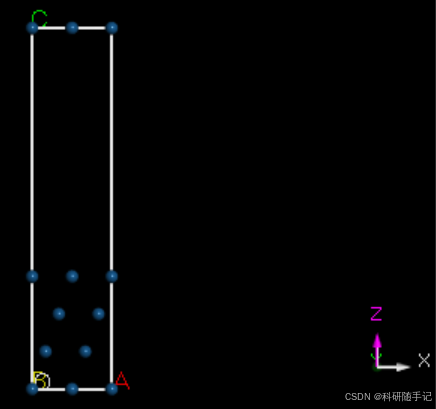
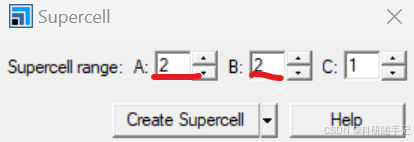
2.1.2 构建slab结构并扩胞
我们要构建一个2x2的supercell,第一种方式是先进行结构优化再扩胞,也第二种方式是先扩胞再进行结构优化。一般来说,当建立的超胞比较大时,为了节省时间优先选择第一种结构优化方式。考虑到此次建模较为简单,结构优化比较快,所以先扩胞再优化。
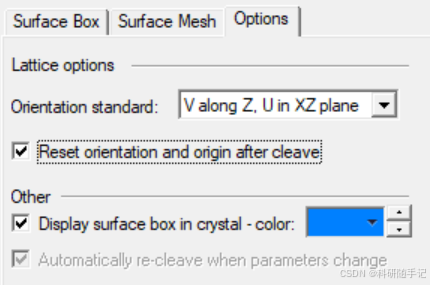
 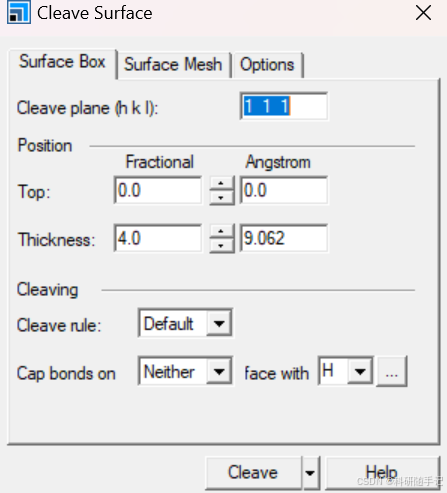 |
  |
  |
 |
  |
 |
2.1.3 构建CO分子
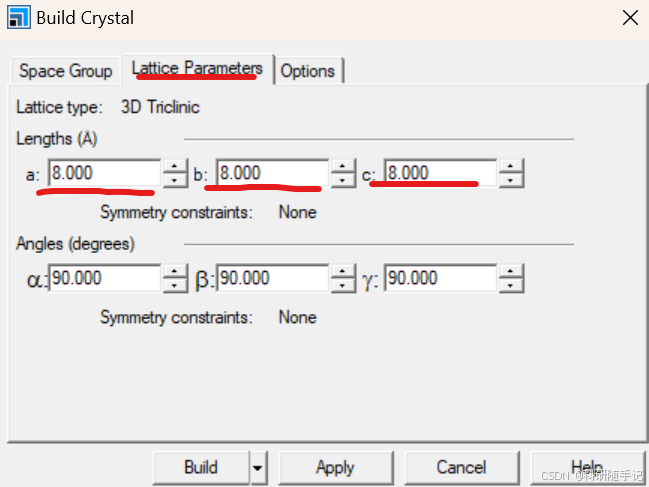
新建一个3D画布New→3D Atomistic Document→Build Crystal建一个8*8*8的格子→Build里Add Atoms(注意option中的坐标选择Cartesian)→依次添加C原子和O原子(C与O之间的键长为1.1283,所以O的z坐标为1.1283)
  |
  |
 |
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 313
313

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









