






作者,Evil Genius
315打假日
2月26日获知家里电信诈骗,到今日过去了17天,从一开始的震惊,到冷静也仅用了3天, 我特别感谢那些帮助了我的人,很多人无偿捐助了我。
很多人都还是学生,将来都会走向社会,进入岗位,其中有一些人也会遇到很大的挫折,我希望大家遇到挫折的时候可以想起我,我这么倒霉的情况下,依然要相信,生活还是很美好的,大多数人的挫折比起我来,也就不再是挫折了。
今天我们来分享一个关于蛋白活性推断的内容,最近一段时间因为一篇文章的发表,运用基因表达来推断蛋白活性,文章在Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages,杂志 Cell,顶刊,其中就用到了单细胞转录组数据来推断蛋白活性,其中用到的软件是viper,2021年5月的一个软件,值得关注。
推断原理
VIPER(Virtual Inference of Protein-activity by Enriched Regulon analysis)算法允许在单个样本的基础上,从基因表达谱数据计算蛋白质活性推断。它利用最直接受特定蛋白质调控的基因表达,如转录因子(TF)的靶标,作为其活性的准确推断手段。
viper实现了一种专门用于估计调控活性的算法,该算法考虑了调节子的作用模式, regulator-target gene相互作用的可信度和每个靶基因调控的多效性。
VIPER在这个包中提供了两种推断方法:多样本版本(msVIPER)设计用于基于多个样本或表达谱的基因表达特征,以及单样本版本(VIPER),它在逐个样本的基础上估计相对蛋白质活性,从而允许将典型的基因表达矩阵(即多个样本中的多个mRNA)转换为蛋白质活性矩阵,表示每个样本中每个蛋白质的相对活性。
看一下实例代码
安装,其中bcellViper提供了示例数据和需要的调控网络作为参考
if (!requireNamespace("BiocManager", quietly=TRUE))
+ install.packages("BiocManager")
BiocManager::install("mixtools")
BiocManager::install("bcellViper")
BiocManager::install("viper")
Getting started
library(viper)
Generating the regulon object
需要输入两个文件
- gene expression signature
- an appropriate cell context-specific regulatory network. This regulatory network is provided in the format of a class regulon object
调控文件通常是由 ARACNe的输出文件产生的,我们来看一下分析过程:
-
由于ARACNe的资源消耗问题,所有对于单细胞数据针对每个cluster进行计算
-
为了生成准确的、鲁棒性好的ARACNe network,ARACNe需要输入表达矩阵中细胞的大部分转录结构相同的数据。对于单细胞转录组数据而言,这需要在生成ARACNe network之前将数据中的细胞进行clustering。这个cluster可以通过多种方式获取:任何一种用于单细胞聚类分群的方法都可以,也可以是简单的通过前几个主成分进行的简单聚类分群。PISCES包中的Clustering方法有:Partition Around Medioids (PAM), Multi-Way K-Means, and Louvain with Resolution Optimization。PISCES软件使用的是基于Seurat与PISCES R Package 对数据进行的two step optimize resolution cluster:所有的clustering step均分两步完成。Seurat中FindNeighbors与FindClusters函数使用的是Louvain算法,这种算法的缺陷是会导致过度分群。因此,在0.01 ~ 1.0的分辨率(resolution)范围内进行聚类以0.01为间隔,并在每个分辨率值上评估聚类质量,以在此范围内选择最佳的聚类方式。对于每个分辨率值(resolution),将cluster中的细胞数再次取样至1000,并计算这1000个细胞及其cluster标签的silhouette score。对于基因表达数据,Pearson correlation被用于细胞距离矩阵(就是用CorDist函数算出来的dist.mat)被用于计算silhouette score。对于VIPER推断出来的蛋白活性数据,VIPER包中的ViperSimilarity函数计算出的distance metric被用于计算silhouette score。这个过程会针对这1000个细胞随机进行100次,然后得出一个针对一个resolution的mean and standard deviation of average silhouette score。选择使平均silhouette score最大化的最高resolution值作为对数据进行聚类而不过度聚类的最佳resolution。
Clustering完成后就可以产生meta-cells用于输入ARACNe:将cluster中距离最近的10个细胞的reads相加后进一步re-normalizing,生成一个具有250个sample的矩阵用于后续ARACNe(这个地方操作起来有点复杂,资源可以的话将所有细胞输入进行计算)。
如果在数据集中的不同细胞类型已经进行了定义和注释,那么cell-type specific networks可以基于细胞注释得出。然而,由于无监督下的(无细胞定义与注释)PISCES 计算可以进一步确认实验的设计是否有问题并可能进一步得出新的生物学发现,因此推荐无监督下的PISCES 计算。这里大家就用定义好的Seurat分群就可以了。
# Seurat clustering
sce.combined.sct <- RunPCA(sce.combined.sct, verbose = FALSE)
sce.combined.sct <- RunUMAP(sce.combined.sct, dims = 1:30, verbose = FALSE)
sce.combined.sct <- FindNeighbors(sce.combined.sct, dims = 1:30, verbose = FALSE)
sce.combined.sct <- FindClusters(sce.combined.sct, method = "igraph" ,verbose = FALSE)
这样的话,抽取每个cluster的矩阵进行下游推断即可,如果cluster的细胞数过多,则需要下采用或者合并。
ARACNe-network generation
从这里开始ARACNe-AP的教程,这部分内容可以基于windows平台的Terminal或者linux平台完成,这里使用的是linux平台,方法大同小异。
- 先下载基于ARACNe-AP作者放在Github上的ARACNe-AP压缩包,将解压后的文件上传至linux服务器并放在后续ARACNe工作目录下并解压;
- 分别下载并安装JAVA和ANT,并设置相应环境变量
下载JAVA-JDK:https://www.oracle.com/java/technologies/downloads/#java8
下载ANTs:https://ant.apache.org/bindownload.cgi - Xshell或MAC-Terminal登录linux服务器
mkdir JAVA_JDK#创建JAVA安装位置
mkdir ANTs #创建ANTs安装位置
- 将下载好的JDK与ANTs安装文件分别传送至服务器的“/JAVA_JDK”与“/ANTs”文件夹中
- 安装软件:分别进入到/JAVA_JDK”与“/ANTs”文件夹中,解压文件
tar -zxvf /your/pathway/to/apache-ant-1.9.14-bin.tar.gz
tar -zxvf /your/pathway/to/jdk-8u131-linux-x64.tar.gz
-
配置环境文件,进入到个人目录下
vi .profile #使用文本编辑器打开并编辑“.profile”文件 -
分别为JAVA与ANT增加环境变量,编辑一下内容加入到“.profile”文件
export JAVA_HOME=/YOUR/PATHWAY/TO/jdk1.8.0_211
export CLASSPATH=$:CLASSPATH:$JAVA_HOME/lib/
export PATH=$PATH:$JAVA_HOME/bin
export ANT_HOME=/YOUR/PATHWAY/TO/apache-ant-1.9.15/
export PATH=$ANT_HOME/bin:$PATH
- 进入ARACNe工作目录,检查JAVA与ANT是否安装成功, 如果安装成功输入以下命令会弹出详细版本号
source .profile
java -version
ant -version
- 构建JAVA需要的aracne.jar文件
ant main
- 构建ARACNe-network需要两个文件:表达矩阵(Matrix)与基因列表(list),注意表达矩阵中使用ensembl ID,那么基因列表也需要是ensembl ID, 如果矩阵中使用gene symble,那么基因列表也需要是gene symble。这两个文件都放在ARACNe-AP-master.zip解压后的test文件夹中,运行参数中的文件名一定要与你文件夹中的文件名一致,否则java在调用这个文件的时候会报错
- 表达矩阵:单细胞基因表达矩阵,txt格式;
- 基因列表:使用PISCES作者给的:"tfs-hugo.txt", COTFs-"cotfs-hugo.txt",Signaling Proteins-"sig-hugol.txt",因为是三个基因集(文件可以在PISCES作者的github上下载:https://github.com/califano-lab/PISCES/tree/master/data,这里除了有上述提到的3个基因集,还有一个surface基因集,演示过程没有使用),因此针对每一个cluster的表达矩阵都需要跑三遍ARACNe流程,再将最后的三个文件合并成一个,即得到这个cluster的ARACNe-network 。当然基因集可以合并
#Step1 Calculate threshold with a fixed seed
java -Xmx5G -jar dist/aracne.jar -e 单细胞cluster矩阵txt -o outputFolder --tfs test/tfs.txt --pvalue 1E-8 --seed 1 \--calculateThreshold
#Step2: Run ARACNe on a single bootstrap
java -Xmx5G -jar dist/aracne.jar -e 单细胞cluster矩阵txt -o outputFolder --tfs test/tfs.txt --pvalue 1E-8 --seed 1
#Step3: Run 100 reproducible bootstraps
for i in {1..100}
do
java -Xmx5G -jar dist/aracne.jar -e 单细胞cluster矩阵txt -o outputFolder --tfs test/tfs.txt --pvalue 1E-8 --seed $i
done
#Step4: Consolidate bootstraps in the output folder
java -Xmx5G -jar dist/aracne.jar -o outputFolder --consolidate
#Step5: Run a single ARACNE with no bootstrap and no DPI
java -Xmx5G -jar dist/aracne.jar -e 单细胞cluster矩阵txt -o outputFolder --tfs test/tfs.txt --pvalue 1E-8 --seed 1 \ --nobootstrap --nodpi
#Step6: Consolidate bootstraps without Bonferroni correction
java -Xmx5G -jar dist/aracne.jar -o outputFolder --consolidate --nobonferroni
-
最后在当前工作目录下进入“outputFolder", 里面的”network.txt“就是要的结果,我们将一个矩阵针对tfs-hugo.txt、cotfs-hugo.txt、sig-hugol.txt的三次运行结果合并在一起,并使用文本打开network.txt文件,将其表头“Regulator Target MI pvalue”删除,保存后修改文件后缀为tsv,就可以将其输入RegProcess()函数了。最终得到的文件就是这个矩阵的ARACNe-network,几个矩阵就有几个ARACNe-network。
-
然后将输出结果中的 .adj file和基因表达矩阵,转换成 regulon object.
Protein activity based clustering analysis
构建的ARACNe-network通过RegProcess()函数将每一个cluster的ARACNe-network文件(pisces-1-net-final.tsv)与其相应的表达矩阵基因表达矩阵(MetaCells函数生成的表达矩阵)进行综合并生成一个调节子对象文件(pisces-1-net-pruned.rds)。
RegProcess <- function(a.file, exp.mat, out.dir, out.name = '.') {
require(viper)
processed.reg <- aracne2regulon(afile = a.file, eset = exp.mat, format = '3col')
saveRDS(processed.reg, file = paste(out.dir, out.name, 'unPruned.rds', sep = ''))
pruned.reg <- pruneRegulon(processed.reg, 50, adaptive = FALSE, eliminate = TRUE)
saveRDS(pruned.reg, file = paste(out.dir, out.name, 'pruned.rds', sep = ''))
}
#针对每个cluster(3个为例)
RegProcess('pisces-1-net-final.tsv', mat1, out.dir = 'tutorial/', out.name = 'pisces-1-net-')
RegProcess('pisces-2-net-final.tsv', mat2, out.dir = 'tutorial/', out.name = 'pisces-2-net-')
RegProcess('pisces-3-net-final.tsv', mat3, out.dir = 'tutorial/', out.name = 'pisces-3-net-')
#RegProcess()函数生成的文件保存为rds格式,先使用readRDS()函数读进来。
c1.net <- readRDS('pisces-1-net-pruned.rds')
c2.net <- readRDS('pisces-2-net-pruned.rds')
c3.net <- readRDS('pisces-3-net-pruned.rds')
#使用list()函数构建net.list文件
net.list<-list(c1.net,c2.net,c3.net)
## viper and protein-activity based clustering
## net.list here would be a list of networks generated from ARACNe
sce.combined.sct <- AddProteinAssay(sce.combined.sct)#将Seurat对象里的count矩阵拿出来命名为PISCES,然后放回到Seurat对象里去,并设置为当前默认的active.assay
sce.combined.sct <- CPMTransform(sce.combined.sct)#针对PISCESassay进行CPMTransform()、GESTransform()标准化。
sce.combined.sct <- GESTransform(sce.combined.sct)
sce.combined.sct <- PISCESViper(sce.combined.sct, net.list)#蛋白活性推断
推断出最终的VIPER矩阵(蛋白活性矩阵),就可以对细胞重新进行clustering。VIPER结果通常允许分辨出更小的、转录上更不同的cluster。然后这些cluster可以被用于鉴定master regulator protein。也可以运用蛋白矩阵进行下游的再分析以及更多的个性化分析。
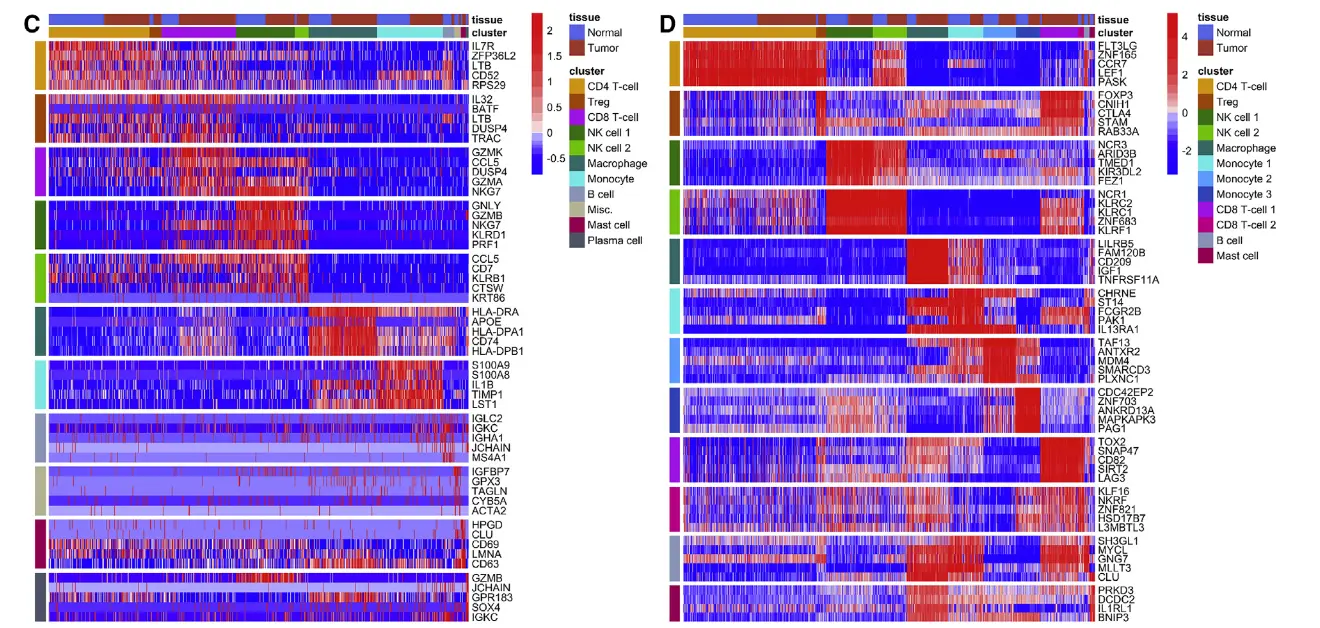
左:差异基因热图;右:差异蛋白表达热图


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









