






07. GO
mkdir 07.GO && cd 07.GO第1步:整合GO注释结果
方法一
整合eggNOG和InterPro中的GO注释结果
发现使用本地化InterProScan的注释结果中没有GO注释
但是使用EBI的工具时是有的(存在报错)
/media/aa/DATA2/bin/go_from_eggNOG_and_interpro.pl /media/aa/DATA/SZQ2/bj/functional_annotation/04.eggNOG/pep70/eggnog_net70/$i.out.emapper.annotations /media/aa/DATA/SZQ2/bj/functional_annotation/05.InterProScan/pep70/$i.interpro.tsv > $i.go1.annot批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "/media/aa/DATA2/bin/go_from_eggNOG_and_interpro.pl /media/aa/DATA/SZQ2/bj/functional_annotation/04.eggNOG/pep70/eggnog_net70/$i.out.emapper.annotations /media/aa/DATA/SZQ2/bj/functional_annotation/05.InterProScan/pep70/$i.interpro.tsv > $i.go1.annot"
done > command.go_from_eggNOG_and_interpro.list
ParaFly -c command.go_from_eggNOG_and_interpro.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "/media/aa/DATA2/bin/go_from_eggNOG_and_interpro.pl /media/aa/DATA/SZQ2/bj/functional_annotation/04.eggNOG/pepmy/eggnog_netmy/$i.out.emapper.annotations /media/aa/DATA/SZQ2/bj/functional_annotation/05.InterProScan/pepmy/$i.interpro.tsv > $i.go1.annot"
done > command.go_from_eggNOG_and_interpro.list
ParaFly -c command.go_from_eggNOG_and_interpro.list -CPU 48方法二
整合eggNOG中的GO注释结果
零基础快速完成基因功能注释 / GO / KEGG / PFAM... - 简书
eggNOG-mapper 大名鼎鼎,是一款非常全面,高效,准确,且一直在更新的软件,对应的,该团队提供了网页接口,任何人可以提交蛋白序列文件,在极短的时间内(一般几分钟)完成基因功能注释,包括:
1.具体功能描述信息
2.Gene Onotoloy注释信息
3.KEGG 注释信息
4.PFAM 注释信息
5.以及其他...
eggNOG-mapper Helper
直接整理 eggNOG-mapper 的结果,输出几个文件,分别满足不同下游分析需要。
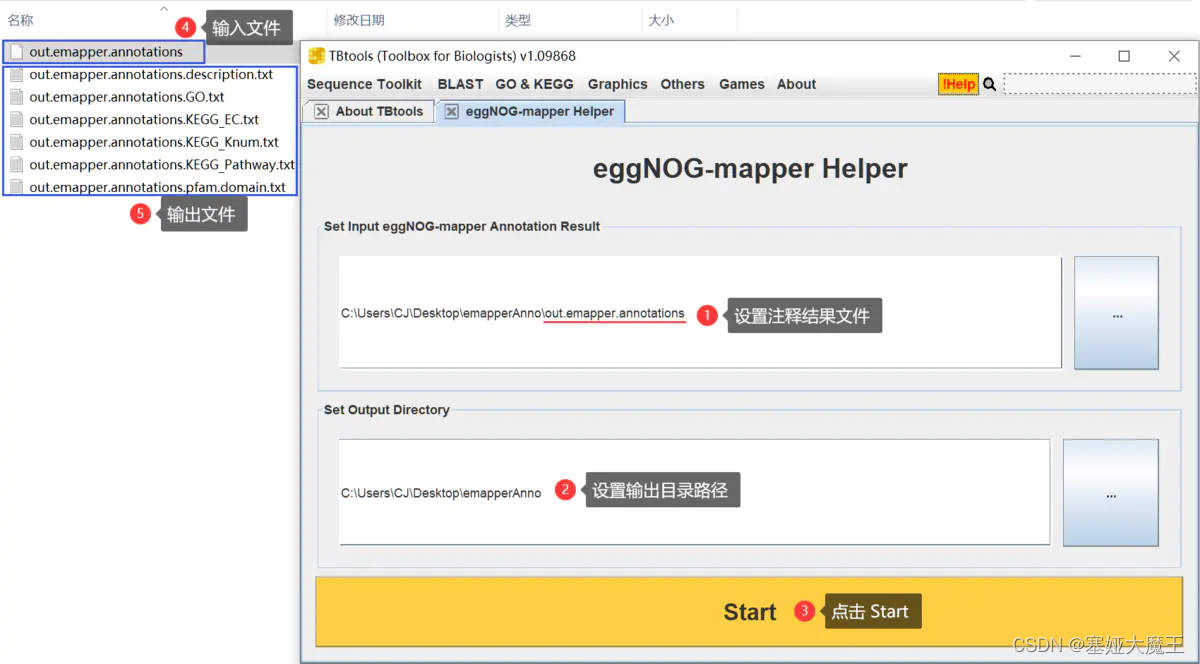
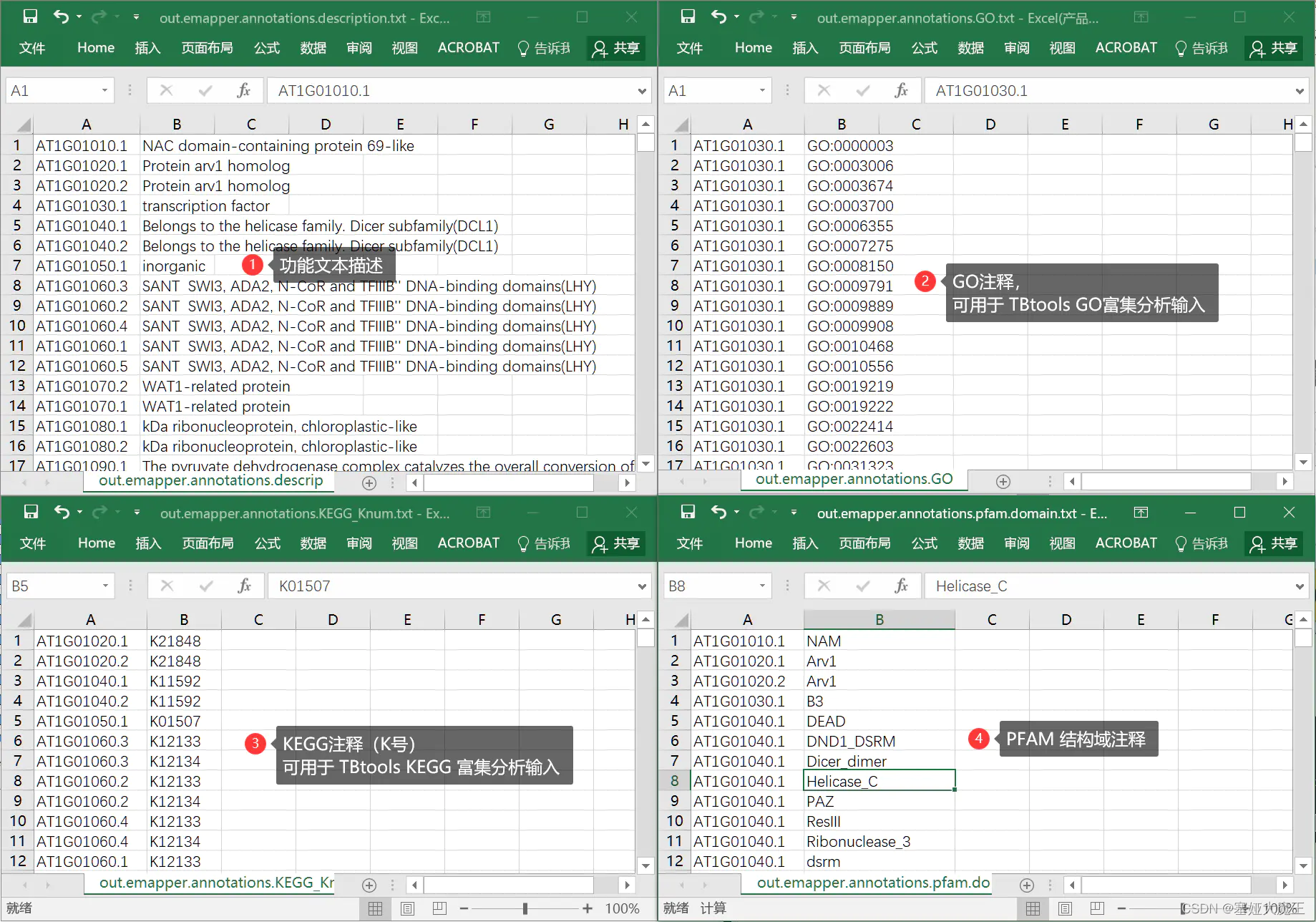
输出文件有四个:
# 对应的功能文本描述
out.emapper.annotations.description.txt
# 对应的是GO注释结果,可直接用于 TBtools GO富集分析,当注释背景文件
out.emapper.annotations.GO.txt
# 对应的是KEGG注释结果,可直接用于 TBtools KEGG富集分析,当背景注释文件
out.emapper.annotations.KEGG_Knum.txt
# 对应的是PFAM结构域注释,注意,这个注释结果是定性的,即有无某结构域,如果一个序列有多个相同结构域,只会显示一个
out.emapper.annotations.pfam.domain.txt四个文件的具体信息,截图可看
注意:这些文件是制表符分隔的文本文件,千万不要保存为 Excel 格式,以免有后续问题!
第2步:go_reducing_go_number.pl
/media/aa/DATA/SZQ2/command/clf/bin/go_reducing_go_number.pl /opt/biosoft/go_class/bin/go.obo $i.go1.annot > $i.go_reduced.annot批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "/media/aa/DATA/SZQ2/command/clf/bin/go_reducing_go_number.pl /opt/biosoft/go_class/bin/go.obo $i.go1.annot > $i.go_reduced.annot"
done > command.go_reducing_go_number.list
ParaFly -c command.go_reducing_go_number.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "/media/aa/DATA/SZQ2/command/clf/bin/go_reducing_go_number.pl /opt/biosoft/go_class/bin/go.obo $i.go1.annot > $i.go_reduced.annot"
done > command.go_reducing_go_number.list
ParaFly -c command.go_reducing_go_number.list -CPU 48第3步:得到go.annot
sort $i.go_reduced.annot > $i.go.annot批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "sort $i.go_reduced.annot > $i.go.annot"
done > command.sort.list
ParaFly -c command.sort.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "sort $i.go_reduced.annot > $i.go.annot"
done > command.sort.list
ParaFly -c command.sort.list -CPU 48第4步:得到GO.txt
下面2种方法选一种使用即可
方法一√
/media/aa/DATA2/bin/gene_annotation_from_table.pl $i.go.annot > $i.GO.txt批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "/media/aa/DATA2/bin/gene_annotation_from_table.pl $i.go.annot > $i.GO.txt"
done > command.gene_annotation_from_table.list
ParaFly -c command.gene_annotation_from_table.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "/media/aa/DATA2/bin/gene_annotation_from_table.pl $i.go.annot > $i.GO.txt"
done > command.gene_annotation_from_table.list
ParaFly -c command.gene_annotation_from_table.list -CPU 48方法二
/media/aa/DATA/SZQ2/go_class/bin/annot2gaf.pl /media/aa/DATA/SZQ2/go_class/bin/go.obo go.annot > gene_association.gaf2
cut -f 2,5,9 gene_association.gaf2 | /media/aa/DATA1/bin/gene_annotation_from_table.pl - > # GO2.txt与GO.txt一样第5步:GO wego分类图(按照goslim_agr.obo方法分了53类)
/opt/biosoft/go_class/bin/annot2wego.pl $i.go.annot > $i.go.wego批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "/opt/biosoft/go_class/bin/annot2wego.pl $i.go.annot > $i.go.wego"
done > command.go.wego.list
ParaFly -c command.go.wego.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "/opt/biosoft/go_class/bin/annot2wego.pl $i.go.annot > $i.go.wego"
done > command.go.wego.list
ParaFly -c command.go.wego.list -CPU 48第6步:得到go_class.tab
/opt/biosoft/go_class/bin/get_Genes_From_GO.pl /opt/biosoft/go_class/bin/go.obo $i.go.wego > $i.go_class.tab批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "/opt/biosoft/go_class/bin/get_Genes_From_GO.pl /opt/biosoft/go_class/bin/go.obo $i.go.wego > $i.go_class.tab"
done > command.go_class.list
ParaFly -c command.go_class.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "/opt/biosoft/go_class/bin/get_Genes_From_GO.pl /opt/biosoft/go_class/bin/go.obo $i.go.wego > $i.go_class.tab"
done > command.go_class.list
ParaFly -c command.go_class.list -CPU 48接下来进行数据整理与画图

















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









