






# 直接百度搜索MetNormalizer,进入Github最简单
##从Github安装Metnormalizer包
if(!require(devtools)){
install.packages("devtools")
}
devtools::install_github("jaspershen/MetNormalizer")
#Github中提供的示例数据,需要下载,不知道数据类型可以下载调整自己的数据
devtools::install_github("jaspershen/demoData")
#其中需要两个文件,第一个是质谱的定量表
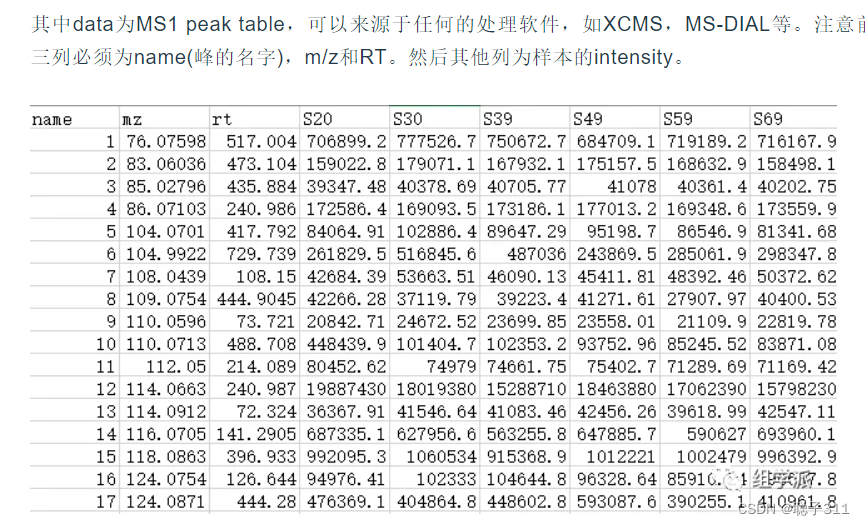
第二个是样品名
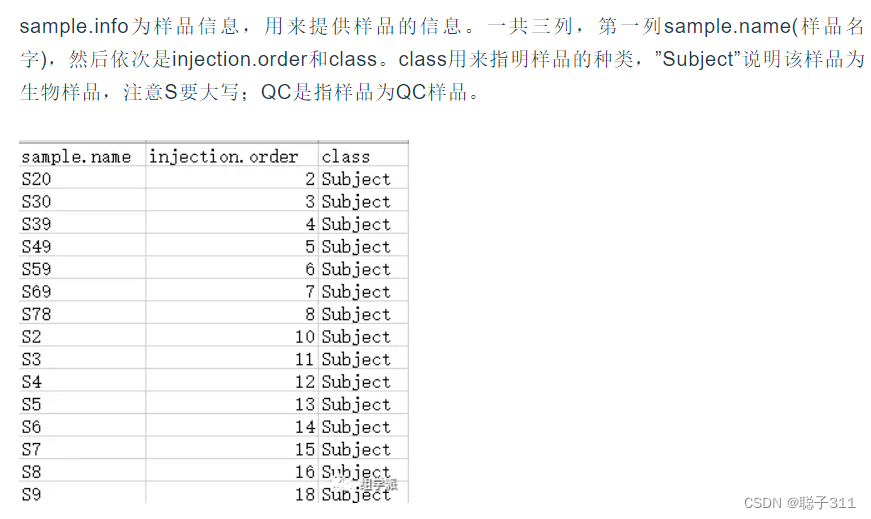
#加载包,导入示例数据
library(demoData)
library(MetNormalizer)
path <- system.file("MetNormalizer", package = "demoData")
file.copy(from = path, to = ".", overwrite = TRUE, recursive = TRUE)
new.path <- file.path("./MetNormalizer")
#直接标准化数据
metNor(
ms1.data.name = "data.csv",
sample.info.name = "sample.info.csv",
minfrac.qc = 0,
minfrac.sample = 0,
optimization = TRUE,
multiple = 5,
threads = 4,
path = new.path
)
















 2037
2037











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









