






目录
二、导入GAPIT(Import library and GAPIT functions run this section each time to start R)
【报错2】“不存在叫‘LDheatmap’这个名字的程辑包” 或 “package ‘LDheatmap’ is not available for this version of R”
写在前面:这是我第一次跑GWAS,所以就把这个过程记录下来啦,有什么不足之处可以指出来~(也参考了一些文章,如果不可以用的话可以联系我删掉)
〇、参考GAPIT网址中的说明文档
https://zzlab.net/GAPIT/gapit_help_document.pdf https://zzlab.net/GAPIT/gapit_help_document.pdf
https://zzlab.net/GAPIT/gapit_help_document.pdf
一、数据准备(以文档中的数据为例)
下载data:http://zzlab.net/GAPIT/GAPIT_Tutorial_Data.zip
(!解压缩之后的数据文件位置一定要与当前工作目录保持一致,可以用一下操作查询或更改路径)
getwd() #查看当前工作目录位置
setwd("目标路径") #如果你不想把文件放在当前目录,可以使用该命令更改路径~二、导入GAPIT(Import library and GAPIT functions run this section each time to start R)
source("http://zzlab.net/GAPIT/GAPIT.library.R")【报错1】显示多个程辑包需要载入
--解决方法:
if(!require(gplots)) install.packages("gplots")
if(!require(LDheatmap)) install.packages("LDheatmap")
if(!require(genetics)) install.packages("genetics")
if(!require(ape)) install.packages("ape")
if(!require(compiler)) install.packages("compiler")
if(!require(grid)) install.packages("grid")
if(!require(bigmemory)) install.packages("bigmemory")
if(!require(EMMREML)) install.packages("EMMREML")
if(!require(scatterplot3d)) install.packages("scatterplot3d")
if(!require(lme4)) install.packages("lme4")
# if(!require(rgl)) install.packages("rgl")
if(!'multtest'%in% installed.packages()[,"Package"]){
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("multtest")
BiocManager::install("snpStats")
}
【报错2】“不存在叫‘LDheatmap’这个名字的程辑包” 或 “package ‘LDheatmap’ is not available for this version of R”
--解决方法
install.packages("LDheatmap", repos='https://mran.microsoft.com/snapshot/2019-02-01/')
library("LDheatmap")不报错之后重新运行一下
source("http://zzlab.net/GAPIT/GAPIT.library.R")
source("http://zzlab.net/GAPIT/gapit_functions.txt")√ 下一步
三、导入文件数据
myY <- read.table("http://zzlab.net/GAPIT/data/mdp_traits.txt", head = TRUE) #Phenotype
myG <-read.table("mdp_genotype_test.hmp.txt", head = FALSE)
myGD=read.table(file="http://zzlab.net/GAPIT/data/mdp_numeric.txt",head=T) #Genotype Data in Numeric Format
myGM=read.table(file="http://zzlab.net/GAPIT/data/mdp_SNP_information.txt",head=T) #Genotype Map for Numeric Format
myKI <-read.table("KSN.txt", head = FALSE) #Kinship Matrix
myCV <-read.table("mdp_population_structure.txt", head = TRUE) #Covariate Variables1、Phenotypic Data
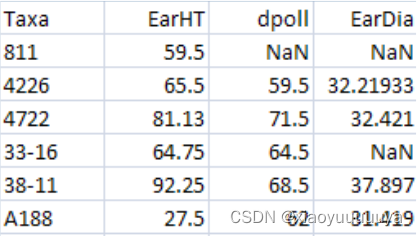
格式如下,第一列必须是Taxa names,其他列是个体表型值,NaN或NA表示缺失的数据,第一行是列标签。
2、Genotypic Data
(1)Hapmap Format
存储序列数据的常用格式,其中SNP信息存储在行中,第一行包含标题标签;单元信息存储在列中,前11列显示了SNP的属性(事实上GAPIT只使用了其中3列:rs列、chrom列、pos列,分别是SNP名称、SNP染色体、SNP的碱基对(bp)位置,余下的列可以用NA填写),其余列显示了每个分类单元在每个SNP上观察到的核苷酸。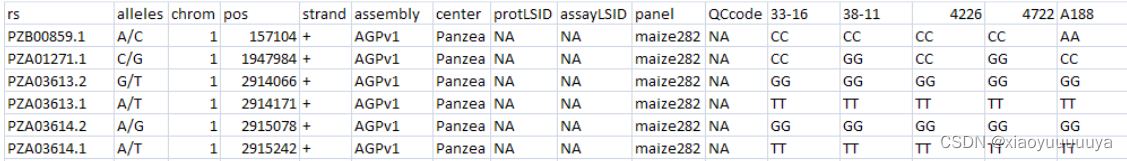
(2)Numeric Format
该格式下需要为GAPIT提供两个单独文件,一个文件包含numeric genotypic data (称为GD文件),另一个文件包含基因组上每个SNP的位置(称为GM文件)。
GD文件中纯合子用0和2表示,杂合子用1表示。0到2之间的任何数值都可以表示估算的SNP基因型。第一行是是SNP names,第一列是taxa names。如下图:

GM(genetic map)文件包含每个SNP的名称和位置。第一列是SNP id,第二列是染色体,第三列是碱基对位置

3、Kinship
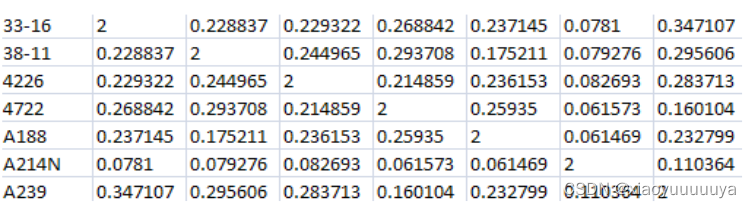
亲缘矩阵文件(KI)被格式化为n × (n+1)矩阵,其中第一列是taxa name,其余是方阵对称矩阵。注意:亲缘矩阵文件的第一行不包含头文件。
4、Covariate variables
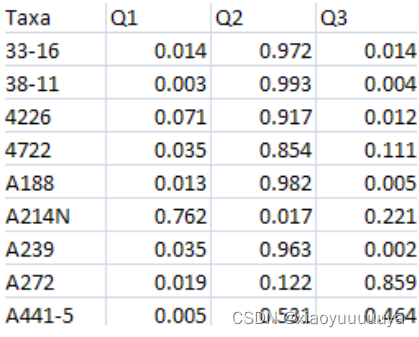
包含协变量的文件(CV)包含population structure(通常称为Q矩阵)等信息。格式与表型文件类似。第一列由taxa names组成,其余列包含协变量值。第一行由column labels组成。第一列可以标记为Taxa,其余列应该是协变量名称。
四、GWAS运行
GAPIT可使用GLM、MLM、MLMM、SUPER、FarmCPU、BLINK等模型,介绍与选择见文档。
GAPIT句法:

GAPIT参数: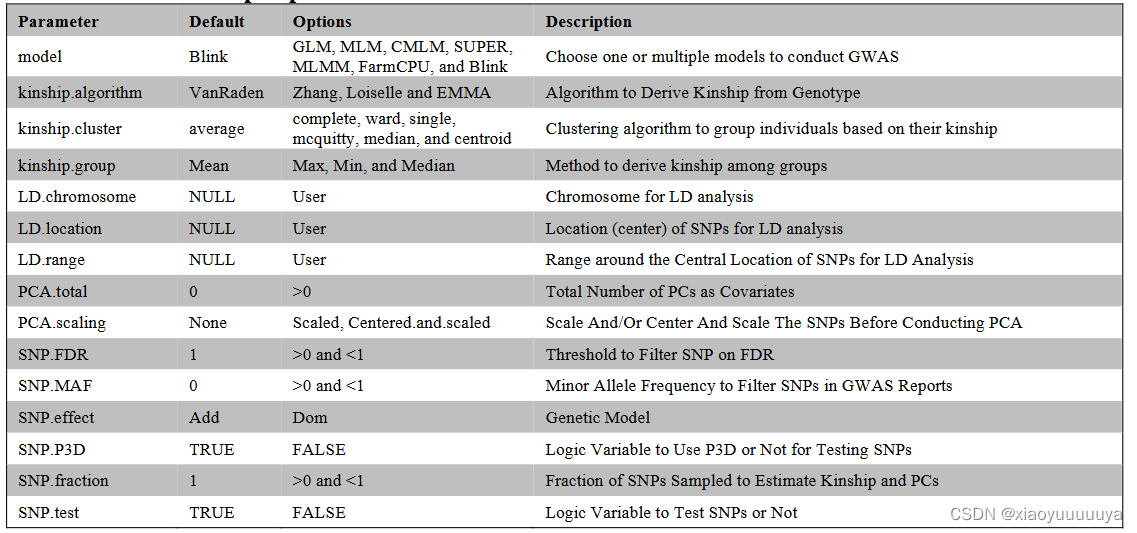
【场景】对于umeric Genotype Format(包含GD文件与GM文件),GAPIT运行命令如下:
myGAPIT=GAPIT(
Y=myY[,c(1,2,3)], #fist column is ID
GD=myGD, #genotypic data文件
GM=myGM, #genetic map文件
PCA.total=3,
model=c("FarmCPU", "Blink"), #FarmCPU、Blink模型
Multiple_analysis=TRUE)其余场景见文档Part 6:Tutorials
五、结果分析
1、表型分布图
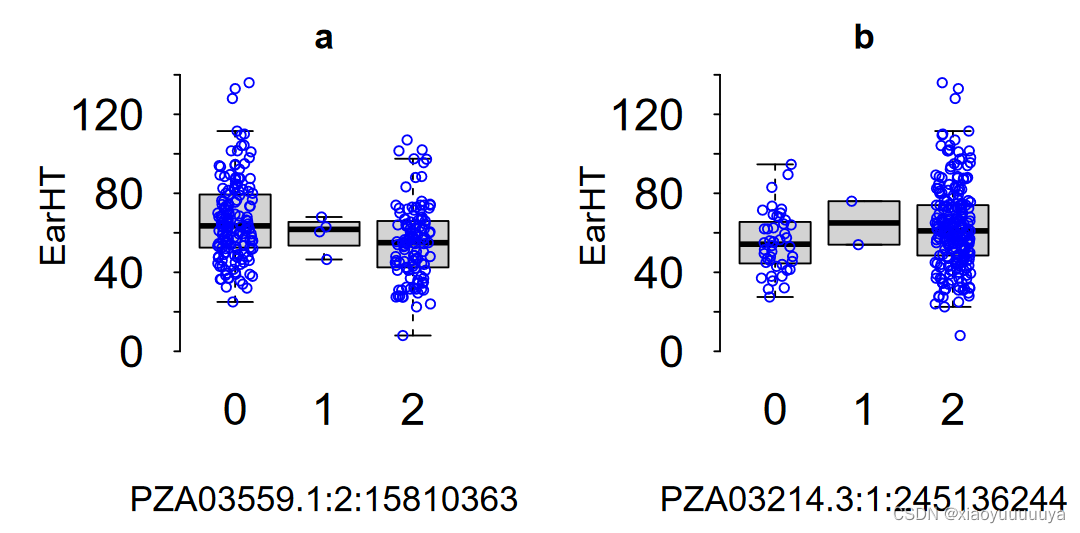
(上图为Blink模型下的EarHT性状)
2、PCA结果
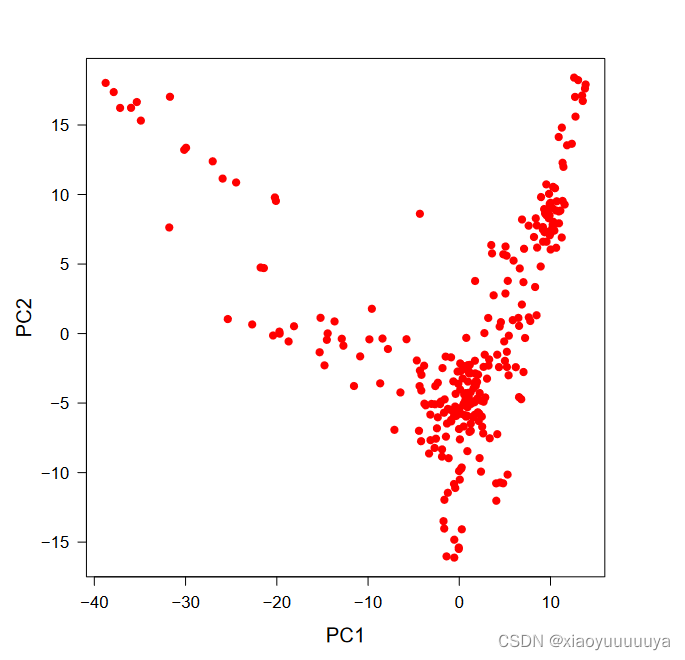
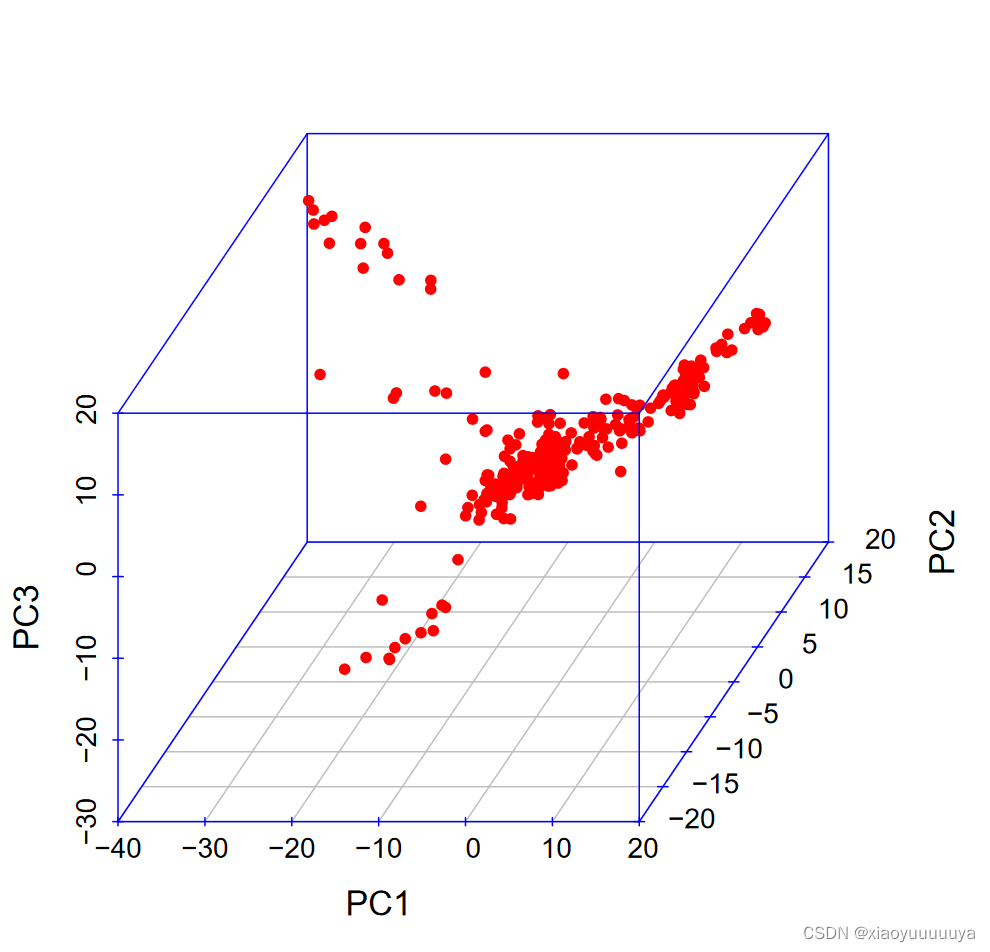
(左:2-D;右:3-D)
3、曼哈顿图
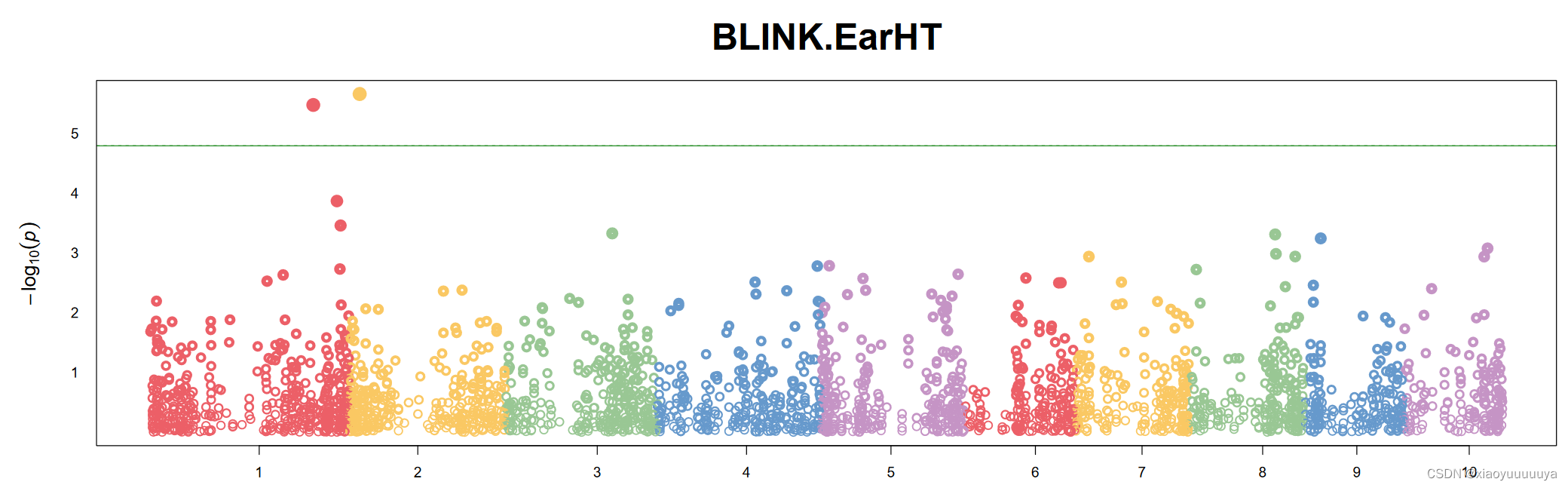
超过阈值的信号峰位置表示此处SNP和表型性状有着很强的相关信号,但不能直接认为这些位点就与表型显著相关的。
这是由于在基因组上引起基因位点的突变有自然突变和遗传漂变,只有自然突变是GWAS研究所关心的,要想排除遗传漂变的影响,需要通过QQ-plot图进一步观察。
(参考文章:如何理解GWAS中Manhattan plot和QQ plot所传递的信息 - 知乎)
4、QQ-plot 图
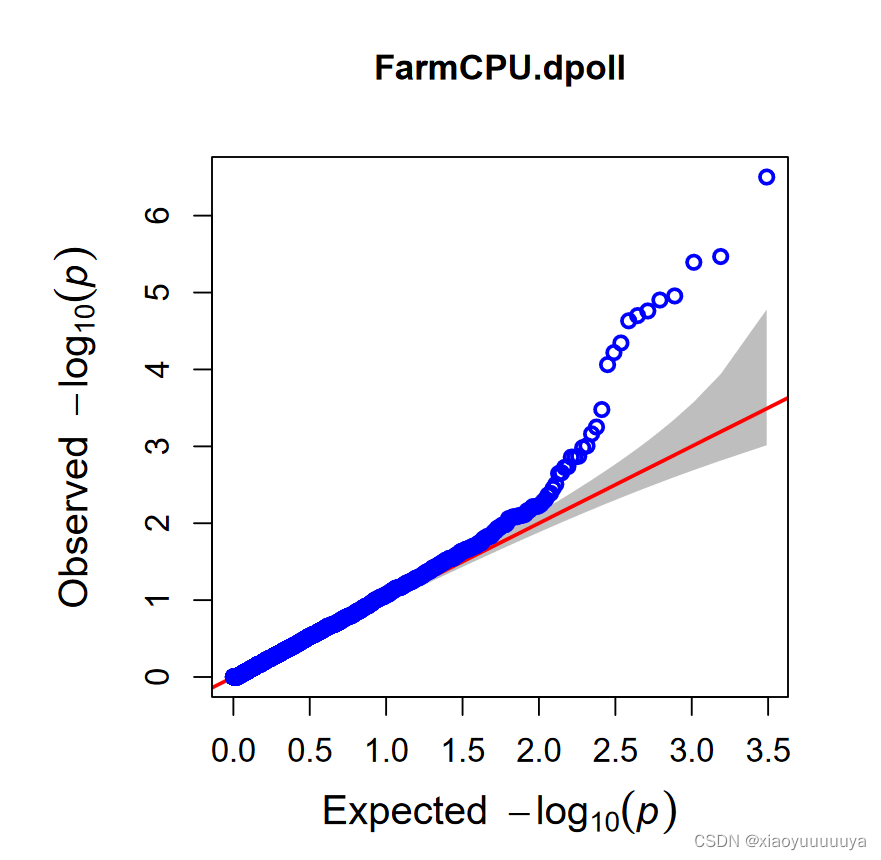
该图形的左下角相关程度低位点中p value的期望值和观察值相等,说明了模型的合理性,右上角翘起来则说明找到了dpoll表型的显著关联位点。
(对QQ-plot结果分析参考QQ-plot图 - 简书)















 2930
2930











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









