






fa文件与fq文件互相转换
今天分享的内容是fasta文件与fastq文件的基本知识,以及通过Python实现两者互相转换的方法。
测序数据公司给的格式通常是fq.gz,也就是fastq文件,计算机的角度来说,生物的序列属于一种字符串,就是一堆字母,这些字母就蕴含了遗传信息。
通过序列拼接将fastq转换为fasta,通过短序列比对将fastq与fasta合并为bam,通过变异检测将bam中突变位点提取出来转换为vcf,这就是上游分析的套路。
fastq文件基本格式
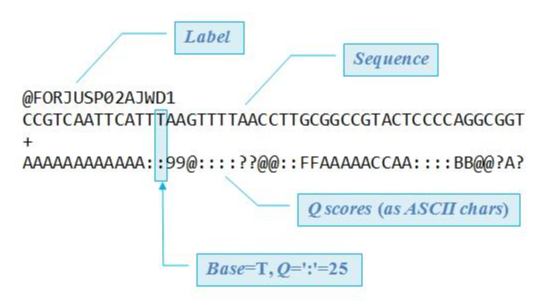
可以看出fq文件包含了更多的信息,比如测序质量,碱基信息等,这些是通过测序仪产生的数据。
fasta文件基本格式
 对比一下可以看出,fa文件主要是两部分,大于号开头的是序列的ID,下一行是序列,相比于fq文件,少了质量信息。
对比一下可以看出,fa文件主要是两部分,大于号开头的是序列的ID,下一行是序列,相比于fq文件,少了质量信息。
将fasta文件转换为fastq文件
分享一个Python脚本实现这个操作:
import sys
fa_f = sys.argv[1]
fq_f = sys.argv[2]
len_max = int(sys.argv[3]) # fq len max: 150bp
with open(fa_f, 'r') as f, open(fq_f, 'w') as pf:
while True:
name = f.readline().strip()
if not name:
break
if name.startswith('>'):
tmp_name = name.strip('>')
read_id = '@'+tmp_name
seq = f.readline().strip()
if len(seq) <= len_max:
read_info = '{rd_id}\n{seq}\n+\n{qual}\n'.format(rd_id=read_id, seq=seq, qual='F'*len(seq))
else:
read_info = ''
cnt = int(len(seq) / len_max)
for i in range(cnt):
tmp_id = read_id + '_' + str(i)
tmp_seq = seq[i*len_max:(i+1)*len_max]
read_info += '{rd_id}\n{seq}\n+\n{qual}\n'.format(rd_id=tmp_id, seq=tmp_seq, qual='F'*len_max)
lseq = len(seq) % len_max
if lseq != 0:
tmp_id = read_id + '_' + str(cnt)
tmp_seq =seq[-lseq:]
read_info += '{rd_id}\n{seq}\n+\n{qual}\n'.format(rd_id=tmp_id, seq=tmp_seq, qual='F'*lseq)
pf.write(read_info)
使用的方法也很简单,把这个脚本保存为xx.py,然后运行并添加三个参数,第一个是原始fasta文件名,第二个是输出文件名,第三个参数是数字,表示每条序列的最大长度,超过该长度的序列将会被切分成多条。
原理解释
刚刚这段Python脚本的功能是将fasta格式的序列文件转换为fastq格式的序列文件,并且可以对序列进行分割,使得每条序列的长度不超过指定的最大长度。
功能:
读取输入的fasta格式的序列文件。 将fasta序列文件中的序列转换为fastq格式。 如果序列长度超过指定的最大长度(len_max),则将长序列分割成多个子序列,每个子序列长度不超过len_max。 将转换后的fastq格式的序列写入输出文件中。
原理:
通过命令行参数传入fasta格式的序列文件路径(fa_f)、要生成的fastq序列文件路径(fq_f)和最大序列长度(len_max)。 使用with open()语句打开fasta序列文件和要生成的fastq序列文件。
逐行读取fasta序列文件,每次读取两行:第一行为序列ID,第二行为序列信息。 对于每条序列,如果序列长度不超过指定的最大长度,则直接转换为fastq格式;否则,将长序列分割成多个子序列,每个子序列长度不超过len_max。
将fastq文件转换为fasta文件
同样,我们也可以使用Python将fq文件转换为fa文件:
import sys
import gzip
fq_in = sys.argv[1]
fa_out = sys.argv[2]
reads_count = sys.argv[3] # if set [-1], means output all reads
with gzip.open(fq_in, 'r') as f, open(fa_out, 'w') as pf:
cnt = 0
while True:
rd_id = f.readline()
if not rd_id or cnt==int(reads_count):
break
seq = f.readline()
tmp = f.readline()
qual = f.readline()
pf.write('>'+rd_id+seq)
cnt+=1
这段Python代码是一个简单的脚本,用于将gzip压缩的Fastq文件(.fq.gz文件)转换为普通的Fasta文件(.fa文件), 下面是代码的原理和作用:
首先,导入了sys和gzip模块,sys用于接收命令行参数,gzip用于解压缩.fq.gz文件。 从命令行参数中获取输入Fastq文件路径(fq_in)、输出Fasta文件路径(fa_out)和要输出的reads数量(reads_count)。
使用gzip.open函数打开输入的Fastq文件,以只读模式打开。使用open函数打开输出的Fasta文件,以写入模式打开。 设置一个计数器cnt,用于记录已经处理的reads数量。
进入一个无限循环,循环中读取Fastq文件中的每个reads信息: 读取reads的ID行(以'@'开头的行)作为rd_id。 读取reads的序列行作为seq。 读取reads的空行(通常为'+')作为tmp。 读取reads的质量信息行作为qual。
将reads的ID和序列信息写入输出的Fasta文件中,格式为>rd_idseq。 计数器cnt加一。 如果读取的reads数量达到指定的reads_count值,则退出循环。 循环结束后,关闭输入和输出文件。
总的来说,将压缩的Fastq文件解压缩并转换为Fasta格式,同时可以根据指定的reads数量控制输出的reads数量。代码中使用了gzip模块解压缩文件,以及文件读取和写入操作,最终实现了Fastq到Fasta的转换。
以上就是今天分享的全部内容,感谢您的阅读,如果感觉有用,欢迎收藏或者转发,您的支持是我更新的最大动力。
参考资料:
https://blog.csdn.net/sinat_32872729/article/details/117353884
https://blog.csdn.net/weixin_46128755/article/details/127947650
https://zhuanlan.zhihu.com/p/77874271
本文由 mdnice 多平台发布
















 5973
5973











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











