









论文解读:《Leveraging the attention mechanism to improve the identification of DNA N6-methyladenine sites》
文章地址:https://academic.oup.com/bib/article-abstract/22/6/bbab351/6359005?redirectedFrom=fulltext#supplementary-data
DOI:https://doi.org/10.1093/bib/bbab351
期刊:Briefings in Bioinformatics(一区)
影响因子:11.622
发布时间:2021年8月28日
Web服务器:http://csbio.njust.edu.cn/bioinf/al6ma/
数据:http://202.119.84.36:3079/al6ma/Data.html
1.文章概述
DNA N6-甲基腺嘌呤是一种重要的DNA修饰类型,在多种生物学过程中发挥着重要作用。尽管DNA的6mA位点预测方法最近取得了进展,但仍有一些挑战有待解决。例如,虽然手工提取的特征是可以解释的,但包含了冗余的信息,这些信息可能会影响模型训练,并且对训练的模型有负面影响。此外,虽然基于深度学习的模型可以自动进行特征提取和分类,但是它们缺乏这些模型所学习的关键特征的可解释性。因此,大量的研究工作都集中在实现深度学习神经网络的可解释性和直观性之间的平衡。本研究利用双向短时记忆技术和自注意力机制分别从DNA序列中提取关键位置信息,建立了两个新的基于深度学习的模型,用于改进N6-甲基腺嘌呤(N6-methyladenine,6mA)位点的预测。两种方法的表现均以模式生物拟南芥(Arabidopsis thaliana)和黑腹果蝇(Drosophila melanogaster)为基准,并作出评估。在两个基准数据集上,LA6mA的AUROC分别为0.962和0.966,而AL6mA的AUROC曲线下面积分别为0.945和0.941。此外,对注意力矩阵进行了深入分析,解释了隐藏在序列中的与6mA位点预测相关的重要信息。
2.背景
表观遗传学被认为是遗传学的一个重要组成部分,它可以在不同水平上表达,包括蛋白质翻译后修饰、 RNA干扰、DNA修饰等。简单地说,表观遗传学通过某些机制导致可遗传的基因表达或细胞表型改变,而不改变序列。作为一种新的表观遗传调控,DNA甲基化在不同的物种中被发现,并被发现与无数的生物过程密切相关,如细胞分化、神经发育和癌症抑制。不同类型的甲基化可以发生在不同位置的修饰。例如,4- 甲基胞嘧啶(4mC)出现在胞嘧啶嘧啶的嘧啶环的第4位置,5- 甲基胞嘧啶(5mC)出现在嘧啶环的第5位置,而6-甲基腺嘌呤(6mA)出现在腺嘌呤环的第6位置。在所有这些以上提到的,4mC和5mC已经广泛研究由于它们的广泛分布。
长期以来,人们一直认为6mA只存在于细菌中,而真核生物中6mA的分布和功能一直不清楚,因为在早期的研究中没有发现。近年来,受益于高通量测序技术的进步和应用,在真核生物中也检测到了6mA。6mA位点可以通过一系列湿法实验室实验方法检测到,这些方法包括但不限于甲基化DNA免疫沉淀测序、毛细管电泳、激光诱导的荧光和 pacbio 单分子实时测序。实验结果提供了丰富的信息,同时也存在费用高、效率低等明显的缺点。6mA位点在基因组中分布不均匀,目前对6mA修饰功能的认识还很有限。因此,通过单核苷酸分解预测6mA位点,并探索目标甲基化位点周围的关键信息,对于角色塑造基因表达的表观遗传调控及其与人类疾病的关联具有重要意义。
目前已经发展了几种真核生物6mA位点预测的计算方法。早期的方法侧重于手工特征的表示和提取,并使用传统的机器学习算法进行预测。例如,chen等人提出了第一个基于机器学习(ML)的方法 i6mA-Pred 用于6mA位点识别,它使用核苷酸化学性质和核苷酸频率作为输入特征,结合支持向量机(SVM)进行训练和预测。i6mA-DNCP使用二核苷酸组成和基于二核苷酸的DNA属性来作为输入,结合bagging 分类器进行预测。iDNA6mA-Rice采用单核苷酸二进制编码进行序列表示,并利用随机森林进行分类。SDM6A 使用五种不同的编码来识别最佳特征集,输入到支持向量机和极随机树分类器。6mA-Finder使用递归特征消除策略从7种序列衍生特征和3种基于物理化学性质的特征中选择最佳特征组。人们经常用手工提取的特征,例如核苷酸的累积频率(ANF)、k-mer频率、伪电子离子相互作用(EIIP)、位置特定的三核苷酸倾向和伪核苷酸组成来表示序列。此外,有些方法还采用了统计模型。例如,MM-6mAPred使用一个一阶马尔可夫模型来识别水稻基因组中的6mA位点。虽然手工特征和机器学习分类器的结合被广泛应用于基因组序列的处理,但不可避免地会存在一定的缺陷。例如,手工制作的特征有信息的冗余,而且非常主观,尽管它们是可以解释的。此外,分类器对于序列中隐藏的信息往往被忽略,使得手工提取的特征难以作为训练分类器的最佳选择。随着深度学习(DL)技术的发展,一些基于序列的端到端算法被用于6mA位点识别。这些方法包括iDNA6mA (5步规则)、SNNRice6mA、DeepM6A、Deep6mA和i6ma-DNC,它们都是基于卷积神经网络以序列的one-hot编码为输入的模型。特别是,i6ma-DNC将DNA序列分解成二核苷酸成分,然后输入到CNN模型,检测N6-甲基腺嘌呤位点。Deep6mA将CNN和LSTM联合预测了6mA位点,发现在不同物种6mA的位点上有相似的模式。
在本研究中,作者提出了两种针对6mA位点的端到端方法:LA6mA和AL6mA。这两种基于序列的方法自动提取序列特征,以DNA序列为唯一输入,区分6mA位点和非6mA位点,从而避免了提取手工特征的繁琐和过度依赖。此外,双向长短时记忆(Bi-LSTM)用于从DNA序列中捕获重要的短程和长程信息,并采用自注意机制捕获序列的位置信息。通过对基准数据的实验,采用LSTM和自我注意机制的不同组合来检验方法的有效性。此外对注意力矩阵进行了详细的分析,包括输入序列的关键位置、注意向量在注意这些关键位置时的变化以及两种模型和两种模式生物注意层的相似性和差异性。作者发现,正样本和负样本在注意力层面上存在的差异,有助于理解为什么这些模型可以做出正确的预测。大量的实验表明,提出的LA6mA和AL6mA方法的竞争性能比其他现有最先进的6mA的预测方法更优异。LA6mA和AL6mA的在线网络服务器已经实现。
3.数据
在这项研究中,DNA的6mA 数据来自两个物种:拟南芥(Arabidopsis thaliana)和黑腹果蝇(Drosophila melanogaster )。原始数据来自PacBio 公共数据库。通过排除序列方差位于识别修饰位点上游10bp和下游5bp之间、预计甲基化水平变异率大于30%的候选基因进一步筛选出来。经过筛选,分别获得了19632个拟南芥(A. thaliana)和10653个黑腹果蝇(D. melanogaster )的6mA位点。选取同数量的非6mA位点作为负样本。每个非6mA位点距离任何相邻的6mA位点至少200个碱基。
作者进一步筛选这些序列,剔除那些含有不确定DNA碱基位点的序列。最后,保留了19616个拟南芥(A. thaliana)正样本和10653个黑腹果蝇(D. melanogaster )正样本。对于每类生物,按为9:1的比例随机分成训练集和独立测试集。

4.方法
4.1 特征表示
采用二进制一热编码方案对输入的DNA序列进行表示: A = [1,0,0,0] ,C = [0,1,0,0] ,G = [0,0,1,0]和 T = [0,0,0,0,1]。这种编码方案使编码矩阵中的元素与输入序列中的碱基对应,便于注意力矩阵/向量的分析。因此,每个长度为L的DNA序列在编码后被转换成大小为L×4的2D矩阵。每个序列的长度为41bp,由每侧20个核苷酸和中心的腺嘌呤位点组成。
4.2 网络结构

LA6mA的骨架如图1A所示。它的双向LSTM层首先连接到编码矩阵,其中两个双向LSTM层的 num_units = 32。然后利用LSTM的每个时间步长连接注意层,权系数矩阵 T∈RL×k 中的参数 k = 32。最后,在完全连接(FC)层之后,注意层被固定并连接到输出端。FC层中的节点数设置为100。
AL6mA的结构如图1D所示。对长度 L的输入序列进行编码,然后直接连接到注意层。注意层后是一个双向LSTM层,其参数如下: num _ units =128、time _ steps = 41。最后,以最后一个时间步长的输出作为最终的预测结果。
值得注意的是,这两个提出的方法不仅仅用于预测潜在的甲基化位点。它们还能够对模型所关注和利用的隐藏信息进行深入的分析,从而做出预测。图1B和图1C描述了注意力矩阵是如何被分析和解释的。
5.结果
5.1 快速识别关键位置
图2展示了AL6mA和LA6mA对拟南芥(A. thaliana)的实验结果,包括随机初始化的注意向量、最终模型的注意向量和注意向量的变化。

图2C和图2F分别显示了AL6mA和LA6mA方法的随机初始注意向量。可以看出,AL6mA的初始注意重量似乎是随机分布在整个序列中(图2C) ,这明显不同于其他。在LA6mA的情况下,它的初始注意力重量几乎均匀分布(图2F)。当输入序列被输入到一个初始化的LA6mA模型时,LSTM层提取序列的特征,然后传递到注意层,这就是为什么提取的初始注意向量中的值分布均匀的主要原因。
图2A和图2D分别显示了AL6mA和LA6mA的最终注意向量。不管最初注意力的分布情况如何,最终模型注意力的中心区域的权重明显大于边缘区域的权重。这表明中部地区对最终结果的预测贡献更大。此外,作者还推测,右侧区对预测的贡献大于左侧区,具体来说,[-2,9]区域在注意力权重值方面对预测的贡献最大。从生物学的角度来看,这个区域的突变可能会影响腺嘌呤中心位点甲基化的可能性。此外,由这种突变引起的甲基化改变可能导致异常的生物过程。
为了观察注意向量的变化,在模型优化过程中提取注意权重。结果表明,随着时间的推移,中心区域的值逐渐增大,而边缘区域的值逐渐减小,突出表明模型在优化过程中可以自动聚焦于关键区域。令人惊讶的是,注意力向量在关键位置的变化只出现在几个epochs上,而随着迭代的进行,注意力向量的值不断被微调,最终到达高值。图2B和图2E验证了快速识别与模型预测相关的关键位置。
5.2 不同注意层次的异同
最终的注意力矢量的训练良好的AL6mA和LA6mA对黑腹果蝇(D. melanogaster )显示在图3。

总之,LA6mA方法在连接到FC层之前将注意力层置于特征提取层之后。因此,LA6mA模型的注意层关注的是提取的特征,而不是原始序列。在一定程度上,注意力可能会分散,变得更加抽象。另一方面,AL6mA模型将注意层与输入矩阵直接连接,关注底层信息,促进关键位置信息的发现。
5.3 AL6mA揭示了6mA预测的关键核苷酸

为了便于分析注意机制,作者使用PLogo为序列组中的每个位置生成序列标志表示。具体来说,在拟南芥(A. thaliana)和黑腹果蝇(D. melanogaster )中心的A碱基周围的序列被检测和识别。根据 p 值 < 0.05的统计学意义对基准高度进行调整。如图4所示,在两个数据集中,6mA和非6mA位点周围的DNA序列中富集和贫化的核苷酸有显著差异。
对于训练有素的模型,可以从每个输入序列中提取出大小为L× 4的注意矩阵。从不同的序列输入中得到不同的注意矩阵,注意矩阵中的值反映了模型在进行预测时注意的特定区域。如图5A所示,热图提供了一个矩阵的可视化,其中的数值由深色或浅色突出显示,深色表示注意力矩阵的较大值,而浅色则相反。


DNA序列中广泛存在的短核苷酸,被认为具有功能性作用,称为DNA序列模体。在这种情况下,除了关键区域的一个单一序列,作者基于一组样本也分析了结果。具体来说,所有的测试样本被输入训练有素的AL6mA模型,并相应地提取所有的注意矩阵。然后,选择TP和TN样本的注意矩阵计算注意矩阵的均值。平均注意力矩阵可以进一步映射到序列,以表示序列中不同位置提供的信息量。图5B、图5C和图6分别显示了拟南芥(A. thaliana)和黑腹果蝇(D. melanogaster )的结果。

表2和表3分别列出了拟南芥(A. thaliana)和黑腹果蝇(D. melanogaster )的关键核苷酸。我们考虑了数据上有意义的基数。
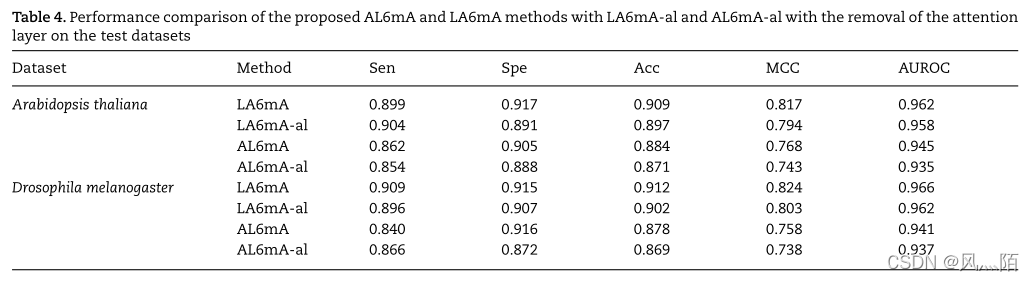
5.4 注意力层对模特表现的影响
针对测试集
去注意层的带al
5.5 提出模型的5倍交叉验证表现

5.6 与现有方法的性能比较



DeepM6A是一个具有315 481 个参数的深度卷积网络,而所提出的AL6mA和LA6mA只有138 043 个(占DeepM6A的43.76%)和159 235 个(占DeepM6A的50.47%)参数。
6.关键点
- 准确预测DNA的6mA位点对于了解其在多种生物过程中的功能作用具有重要角色塑造。
- 发展了两种新的方法,分别称为LA6mA和AL6mA,利用LSTM自动获取DNA序列中的信息。
- 利用自注意机制有效地捕捉DNA序列中的位置信息
- 深入分析了两种6mA预测模型对拟南芥(A. thaliana)和黑腹果蝇(D. melanogaster )注意力权重的变化、注意层的异同,以及TP和TN样本注意层的差异,解释了支持模型预测的关键信息。
- 搭建了一个在线网络服务器,该服务器可作为预测 dna 6mA位点的有用工具。














 4316
4316











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









