






Multi-omic and multi-view clustering algorithms: review and cancer benchmark

文献翻译自用
摘要
最近的高通量实验方法已被用于收集大型生物医学组学数据集。单组学数据集的聚类已被证明对生物和医学研究是无价的。成本的降低和其他高通量方法的发展现在使多基因组数据的测量成为可能。聚类多组数据有可能揭示进一步的系统级见解,但会带来计算和生物学方面的挑战。在这里,我们回顾了多组学聚类的算法,并讨论了应用这些算法的关键问题。我们的综述涵盖了专门为基因组数据开发的方法,以及机器学习社区为多种数据类型的联合聚类开发的通用多视图方法。此外,利用TCGA的癌症数据,我们执行了跨越十种不同癌症类型的广泛基准,提供了领先的多组学和多视图聚类算法的首次系统比较。结果突出了关于使用单组学与多组学,聚类策略的选择,通用多视图方法的力量以及使用近似值来衡量解决方案质量的关键问题。由于越来越多地使用多组学数据,我们预计这些问题对该领域的未来进展很重要。
引言
深度测序和其他高通量方法在单个实验中测量大量分子参数。测量的参数包括DNA基因组序列(1)、RNA表达(2,3)、DNA甲基化(4)等。每一种这样的数据都被称为“组学”(基因组学、转录组学、甲基组学)。随着成本的增加和技术的成熟,可以获得更大、更多样化的基因组数据集。
计算方法对于分析这类数据是必不可少的。一个基本的分析是聚类——在数据中找到连贯的样本组,这样一组内的样本是相似的,而不同组中的样本是不相似的(5)。这种分析通常是数据探索的第一步。聚类在生物医学研究中有许多应用,例如发现共调控基因的模块和在精准医学背景下发现疾病的亚型(6)。聚类是一个被高度研究的计算问题,被多个科学团体研究,并且存在无数算法用于该任务。
虽然对每个组学分别进行聚类可以揭示数据中的模式,但对同一组样本使用多个组学的集成聚类有可能揭示更精细的结构,而这些结构仅通过检查单一数据类型无法揭示。例如,癌症亚型可以同时根据基因表达和DNA甲基化来定义。需要基于多个组学的聚类有几个原因。首先,多组学聚类可以降低实验噪声和生物噪声对数据的影响。其次,不同的组学可以揭示不同的细胞方面,例如在基因组和表观基因组水平上表现的效应。第三,即使在相同的分子方面,每个组也可以包含其他组中不存在的数据(例如突变和拷贝数)。第四,组学可以代表不同生物体水平的数据,如基因表达和微生物组组成。
机器学习社区独立研究了一个类似于多组学聚类的问题,并将其称为“多视图聚类”(见(7)和“多视图聚类调查”)。多视图聚类算法可用于对多组数据进行聚类。在过去,机器学习社区开发的方法在生物医学数据集分析中被证明是有用的。然而,多视图聚类在生物信息学领域的应用还远远不够。
在本文中,我们回顾了多组学聚类的方法,并在真实的癌症数据上进行了基准测试。数据来源是TCGA(癌症基因组图谱)(8),这是一个包含数千名癌症患者数据的大型多基因组数据库。我们调查了多组学和多视图方法,目的是让计算生物学家了解这些算法。在这篇综述中,我们在机器学习算法的上下文中使用术语视图和多视图,而不是组学和多组学。
最近几篇综述讨论了多组学整合。(9-11)综述了多组学整合的方法,(12)综述了多组学聚类在癌症中的应用。这些评论不包括基准测试,也不关注多视图聚类。(13)仅综述了降维多组学方法。据我们所知,(14)是对多组学聚类执行的唯一基准,但它不包括机器学习方法。此外,我们认为在基准测试中测试的方法并不代表多组学聚类的当前状态。最后,(7)是针对机器学习社区的多视图方法的全面回顾。它不讨论由生物信息学社区开发的算法,也不涵盖生物应用。
多组学聚类方法综述
我们根据其算法方法将这些方法分为几类。早期集成是最简单的方法。它将组学矩阵与多个组学的特征连接成一个矩阵,并对该矩阵应用单组学聚类算法。在后期集成中,每个组学分别聚类,并将聚类解集成为单个聚类解。其他方法试图构建一个包含所有组学的模型,并统称为中间集成。这些方法包括:(i)整合样本相似性的方法,(ii)对不同组学数据集使用联合降维的方法,以及(iii)使用数据统计建模的方法。
我们在这里提出的类别并不明确,并且提出的一些算法适合多个类别。例如,iCluster(15)是一种早期的集成方法,它也使用概率建模将数据投影到较低的维度。这些算法在我们认为最适合的类别中进行描述。
多组学聚类算法也可以通过它们支持的组学集来区分。通用算法支持任何类型的组学数据,因此很容易扩展到新的未来组学。组学特定算法针对数据类型的特定组合量身定制,因此可以利用已知的生物学关系(例如拷贝数和表达之间的相关性)。这两种方法的混合是以一种特定的方式进行特征学习,然后使用通用算法对这些特征进行聚类。例如,人们可以用记录细胞通路中表达的组学来取代基因表达组学,从而利用现有的生物学知识。
在整个综述中,我们使用以下表示法:一个多组数据集包含M组。n是样本数量(对于医疗数据集来说是患者数量),是第m组中的特征数量,Xm是包含第m组测量值的n x
矩阵。因此
是第m个组中第i个病人的第j个特征的值。
为特征总数,X为所有Xm个矩阵串联形成的n × p矩阵。在整个论文中,对于矩阵A,我们使用
来表示它的转置,并且与Xm符号一致,我们使用
来表示矩阵索引(而不是矩阵幂)。选择其他符号是为了遵循原始出版物和通用惯例。
图1图形化地总结了多组学聚类的不同方法。表1给出了这里审查的方法的汇总表。
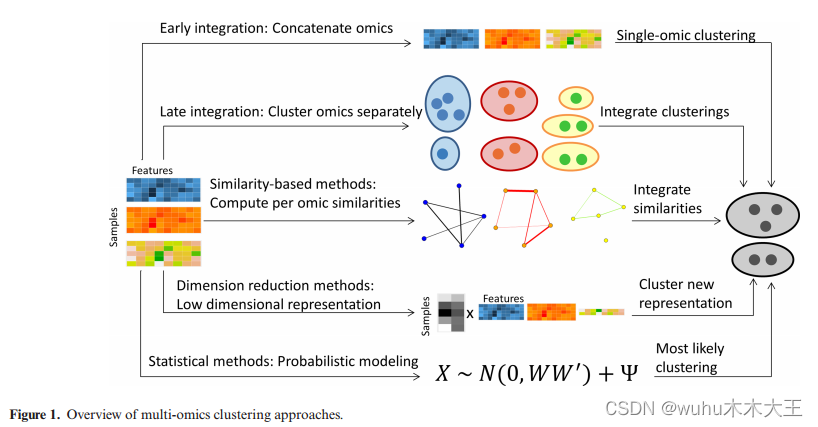
早期的集成
早期集成是一种首先连接所有组矩阵,然后在该连接矩阵上应用单组聚类算法的方法。因此,它可以使用现有的聚类算法。然而,这种方法有几个缺点。首先,如果没有适当的规范化,它可能会赋予具有更多特征的组学更多的权重。其次,它没有考虑不同组学中数据的不同分布。最后,它增加了数据维度(特征的数量),即使在一些单基因组数据集中,这也是一个挑战。在应用专门为多组学数据设计的早期集成算法时,或者在连接矩阵上运行单组学方法时,必须解决这些缺点。对不同组学中的特征进行归一化处理有助于处理不同的分布,并且可以使用特征选择来降低维数,从而给不同组学一个平等的优先机会来影响结果。
处理高维数据的另一种方法是使用正则化,即在问题中添加额外的约束以避免过拟合(76)。具体来说,LASSO(最小绝对收缩和选择算子)正则化创建的模型对模型具有非零影响的特征数量很少(77),并且核范数的正则化通常用于诱导数据稀疏性。实际上,iCluster(15)使用LASSO正则化(将在后面的章节中讨论),而LRACluster使用核范数正则化(将在本节中讨论)。虽然可以使用早期集成应用任何聚类算法,但我们在这里重点介绍专门为此任务开发的算法。
LRACluster(16)使用概率模型,其中数字、计数和二进制特征的分布由样本的潜在表示β决定。例如其中
与Xm具有相同的维数。通过在其核范数上添加正则化,鼓励潜在表示矩阵具有低秩。算法的目标函数为−log(模型的似然)+
·|β|*,其中β为所有βm矩阵的串接,|·|*为核范数。该目标是凸的,并提供了一个全局最优解,该解使用快速梯度上升算法找到。随后使用k-means对β进行聚类。这使用4种不同的组学方法分析了11种癌症类型的泛癌症TCGA数据,并进一步发现这些癌症类型中的亚型。
在(17)中,所有组学都被连接到矩阵X上,算法最小化以下目标:||XW + 1n−F||2+ γ ||W||G1。W是一个p × k的投影矩阵,F是一个n × k的簇指示矩阵,使得
F = Ik, 1n是长度为n的1的列向量,b是维数为k的截距列向量,
是一个标量。因此,该算法寻求一种线性变换,使投影数据尽可能接近聚类指标矩阵。该指标矩阵随后用于聚类。正则化项使用G1范数,它是与特定集群和视图相关的W项的l2范数,对所有视图和集群求和。因此,对聚类结构没有贡献的特征将在W中被分配低系数。
交替优化
关于两种观点整合的早期研究是在(78)中进行的。这项工作使用一种称为协同训练的方法提高了具有两个视图的半监督数据的分类精度,并启发了其他人分析多视图数据。第一次尝试执行多视图聚类是(18)。本文将期望最大化(EM)和k-means这两种常用的单组聚类算法应用于多视图聚类。EM和k-means都是迭代算法,每次迭代都会改进目标函数值。建议的多视图版本在每次迭代中以交替的方式针对不同的组执行优化。这种方法失去了收敛的理论保证,但被发现优于单独使用每个视图的算法,以及对两个视图的连接矩阵进行聚类的朴素早期集成方法。有趣的是,(18)报告了改进结果,在单视图数据集上随机拆分以模拟多视图数据。这是使用多视图改进聚类的第一个证据,也是多视图算法在聚类单视图数据中的实用性的第一个证据。虽然这项工作非常有影响力,但其他初步的多视图聚类方法(例如(22,31))在已知金标准的数据集上取得了更好的结果。
后期集成
后期集成是一种允许在单组数据上使用现有单组聚类算法的方法。首先,使用单组算法对每个组进行单独聚类。每个组可以使用不同的算法。然后,对不同的聚类进行整合。后期集成的优势在于任何聚类算法都可以用于每个组。因此,可以使用已知在特定基因组上工作良好的算法,而不必创建统一所有这些算法的模型。然而,如果在集成阶段只使用聚类解决方案,我们可能会分别丢失每个组中较弱的信号。
将COCA(19)应用于泛癌TCGA数据,研究来自不同组织的肿瘤如何聚类,以及获得的聚类是否与起源组织匹配。该算法首先将每个组分别聚类,使得第m个组有个簇。样本i对于组学 m的聚类被编码成一个长度为
的二进制向量
,其中如果i属于聚类j, vim(j) = 1,否则为0。将所有组学中的vim向量串联起来,得到样本i的二进制聚类指标向量。这些指标向量的n × c二进制矩阵B,其中c =
cm,用作一致性聚类的输入(79),以获得样本的最终聚类。或者,在(20)中提出了一个基于概率潜在语义分析(Probabilistic Latent Semantic Analysis, 80)的模型用于聚类B。这两种方法允许在每个单个组上使用任何聚类算法,因此在特定组学使用已知方法时具有优势。此外,只有在原始基因组数据不可用时,才可以在集群解决方案中使用它们。
PINS(21)通过检查不同组学的连接矩阵来整合集群。每个这样的矩阵Sm都是一个二进制的n x n矩阵,其中,如果患者i和j聚集在组m中,则 = 1,否则为0。对这些Sm矩阵进行平均得到单个连通性矩阵,然后根据不同的Sm矩阵是否高度一致,使用不同的方法对其进行聚类。测试获得的聚类是否可以进一步分成更小的聚类。为了确定每个组和集成聚类的聚类数量,通过添加高斯噪声对数据进行扰动,并选择聚类的数量,使结果聚类对扰动具有鲁棒性。与之前提出的后期积分方法不同,PINS需要原始数据,而不仅仅是每个组的聚类,因为它对数据执行扰动。
多年来开发了几种集成聚类方法,并在(81)中进行了综述。虽然它们最初不是为此目的开发的,但它们也可以用于后期的多组学聚类。
基于相似性的方法
基于相似度的方法使用样本之间的相似度或距离来聚类数据。这些方法分别计算每个组中样本之间的相似性,并且在这些相似性的整合方式上有所不同。集成步骤只使用相似值。由于在当前的多组数据集中,样本数量远小于特征数量,因此这些算法通常比考虑所有特征同时进行集成的方法更快。然而,在这种方法中,用原始特征来解释输出可能会更加困难。基于相似性的方法的另一个优点是,它们可以很容易地支持不同的组类型,包括分类和有序数据。每个组学只需要一个相似性度量的定义。
1.谱聚类推广
谱聚类(82)是一种广泛使用的基于相似性的单视图数据聚类方法。单视图光谱聚类的目标函数为maxxtrace (Ut LU) s.t。Ut U = I,其中L为维数n × n的相似矩阵的拉普拉斯式(83),U为维数n × k,其中k为数据中的聚类个数。直观地说,这意味着彼此相似的样本在u中具有相似的行向量。这个问题可以通过取L的k个第一个特征向量来解决(使用归一化和非归一化图拉普拉斯的版本之间的细节有所不同),并用k-means等简单算法对它们进行聚类。谱聚类目标被证明是图中离散归一化切割的松弛,为聚类提供了直观的解释。几种多视图聚类算法是谱聚类的推广。
对两个视图的早期扩展通过使用两个视图的相似性计算一个新的相似性矩阵来执行聚类(22)。用W1和W2表示两个视图的相似矩阵。那么积分相似度W,定义为W1W2。在分块矩阵上进行谱聚类
这相当于在拉普拉斯Lm上进行谱聚类。然后,通过改变样本对聚类的分配,以贪婪的方式进一步改进得到的聚类,同时直接关注离散的归一化切割目标,而不是连续的光谱聚类目标。
Li(25)建议运行时间优于(23)。不是查看所有样本的相似性矩阵,而是通过对所有组学的连接运行k-means并选择聚类中心来计算一小组“代表性”向量(称为突出点)。然后计算数据中所有这些样本与其最近的5个显著点之间的相似性矩阵。用Wm表示第m组的相似矩阵,设Zm是它的归一化使得行和为1。这些矩阵的维数是n ×突出点的个数。接下来,矩阵
上述方法在几个方面有所不同。(23)允许每个组具有不同的低维表示,并有一个参数来控制这些表示的相似程度之间的权衡,以及如何在Um中保持原始数据的相似性。因此,它允许表达不假设组学具有相同相似性结构的情况(例如,两个样本可以在一个组学中相似,但在另一个组学中不同)。另一方面,Chikhi(24)假设相同的相似结构,在这种情况下,其贪心优化步骤可以得到改进的解。(25)可在样本量特别大的情况下使用。
Zhou和Burges(26)将相似矩阵视为网络,并研究了这些网络上的随机漫步。随机漫步定义了每个网络上的平稳分布,它捕获了其相似模式(84)。由于平稳分布比原始的相似度度量噪声更小,Zhou和Burges(26)转而使用它们来整合网络。Xia(27)也研究了网络上的随机游走,但认为每个网络中的平稳分布仍然可能是有噪声的。相反,作者计算了一个共识转移矩阵,它与每个经济转移矩阵的总距离最小,并且秩最小。随机漫步与谱聚类高度相关;在谱聚类中使用图的拉普拉斯算子的一种归一化变体会得到一个解,其中随机行走很少在聚类之间交叉(82)。这些基于随机行走的方法目前与其他谱聚类方法竞争。
2.相似网络融合
SNF (Similarity Network Fusion)首先为每个组分别构建一个相似网络(28,29)。在每个这样的网络中,节点都是样本,边缘权重衡量样本的相似度。然后使用基于消息传递的迭代过程将网络融合在一起(85)。样本之间的相似性在每个节点与其k个最近邻之间传播。
更正式地说,用W(m)表示第m组的相似矩阵。初始,所有样本之间的转移概率矩阵定义为:

在(29)中,SNF被用于TCGA中几种癌症亚型的基因表达、甲基化和miRNA表达数据。除了划分图以获得癌症亚型外,作者还表明融合网络可以用于其他计算任务。例如,他们展示了如何拟合Cox比例风险(86),这是一个预测患者预后的模型,具有一个约束,使得综合网络中的类似患者具有相似的预测预后。
3.rMKL-LPP
核函数隐式地将样本映射到一个高维度(可能是无限的),并且可以有效地测量该维度中样本之间的相似性。多核学习使用多个核(相似度度量),通常通过线性组合它们,并且在监督分析中经常使用。(30)开发了rMKL-LPP(正则化多核学习与局域保持预测),它在无监督环境中使用多核学习。该算法对输入组学进行降维,使每个样本与其最近邻居之间的相似性(使用多个核定义)保持在低维上。这个表示随后用k-means聚类。rMKL-LPP允许使用不同的内核函数,甚至每个组使用多个内核。在优化问题中加入正则化项以避免过拟合。作者在TCGA中的五种癌症类型上运行了该算法,并表明每个基因组使用多个核提高了聚类的预后价值,并且正则化提高了鲁棒性。
基于降维的方法
基于降维的方法假设数据具有固有的低维表示,这种低维通常对应于簇的数量。我们观察到的视图都是从低维数据到高维数据的转换,不同视图之间转换的参数不同。这个一般公式由(31)提出,它建议最小化M mwml(Xm, fm(B)),其中B是一个维数为n × p的矩阵,fm是参数化变换,
是不同视图的权重,l是损失函数。进一步给出了用矩阵乘法给出调频变换时的优化算法。即fm(B) = BPm, l是应用于Xm−BPm的Frobenius范数的平方。一旦计算出B,就可以对其应用单组聚类算法。这个一般框架被广泛使用。由于通常假定变换是线性的,因此许多降维方法都是基于矩阵分解的。降维方法适用于实值数据。将这些方法应用于离散二进制或计数数据在技术上是可行的,但通常不合适。
线性降维方法的一个优点是,它们为每个聚类的主要特征提供了一些解释。例如,在刚才介绍的一般框架中,Pm矩阵中的每个条目可以被认为是集群中一个特征的权重。这种解释在基于相似度的方法中是缺失的,这种方法在计算样本之间的相似度时忽略了原始特征。因此,当需要在聚类和特征之间建立关联时,降维方法可能是有用的。
1.JIVE
(32)假设每个组的变异可以划分为所有组之间的联合变异,以及组特异性变异:Xmt = Jm + Am + Em,其中Em为误差项。设J和A分别是连接的Jm和Am矩阵。模型假设JAt = 0,即关节与组的特异性变异不相关,且每个组的rank(J) = r, rank(Ai) = ri,则每个组的结构和总关节变异都是低秩的。为了使不同组的权值相等,将输入组矩阵归一化为具有相等的Frobenius范数。增加处罚项以鼓励可变稀疏性。该方法应用于多形性胶质母细胞瘤脑肿瘤的基因表达和miRNA数据,并确定了这些组学之间的联合变异。
2.相关与基于相关
两种最广泛使用的降维方法是典型相关分析(CCA)(34)和偏最小二乘(PLS)(45)。给定两个组学X1和X2,在CCA中,目标是找到p1和p2维的两个投影向量u1和u2,使投影数据具有最大的相关性:argmax1,u2 * (X1 u1, X2 u2)。
这些投影被称为第一典型变量,是组学之间最大相关性的轴。第k对典型变量,u1 k和u2 k被发现使得X1u1 k和X2u2 k之间的相关性是最大的,假设新的对与之前的典型变量不相关(即正交)。Chaudhuri等人(87)通过经验证明并表明,如果数据来源于正态分布或对数凹分布,则可以使用典型变量对数据进行聚类。CCA是在一个概率框架中制定的,使得优化解决方案是最大似然估计(88),并进一步扩展到贝叶斯框架(35)。在高维执行CCA的另一个扩展是Kernel CCA(36)。最近开发了一种基于深度学习的CCA方法DeepCCA(37)。不是最大化数据的线性投影之间的相关性,而是将投影作为使用神经网络计算的数据的函数,优化过程优化这些网络的参数。
求解CCA需要对两个组的协方差矩阵进行反转。组学数据通常比样本具有更多的特征,因此这些矩阵是不可逆的。为了将CCA应用于组学数据,并提高CCA结果的可解释性,我们添加了稀疏性正则化(38,39)。
CCA只支持两个视图。一些研究将其扩展到两种以上的观点,包括MCCA(39),它最大化了预测和CCA-RLS之间的成对相关性之和(40)。Luo等人(41)将CCA推广到张量,以支持两种以上的观点。
CCA的另一项工作与组学数据高度相关,在执行降维时研究了特征之间的关系。ssCCA(结构约束稀疏CCA)允许将一个输入组学中的特征之间的已知关系合并到模型中,并强制该视图的ui向量中的条目接近相似的特征。该模型由(42)建立,利用微生物组的系统发生作为特征结构。另一个考虑特征之间关系的模型是在(43)中提出的。在这项工作中,不是定义特征之间的相似性,而是将它们划分为组。执行正则化,以便从模型中删除相关组中的不相关组和不相关特征。最后,Podosinnikova等人在“超越CCA:多视图模型的时刻匹配”中扩展了CCA以支持计数数据,这在生物数据集中很常见。
PLS也遵循线性降维模型,但最大化预测之间的协方差,而不是相关性。更正式地说,给定两个组学X1和X2, PLS计算一个向量序列u1 k和u2 k, k = 1,2,…使得cov(x1u1k, x2u2k)最大,假设u1 k t u1 k = 1, u2 k t u2 k = 1,并且cor(x1u1k, x1u1l) = 0,对于l < k。也就是说,新的投影与以前的投影不相关。即使没有稀疏性约束,PLS也可以应用于具有比样本更多特征的数据。尽管如此,稀疏解决方案仍然是可取的,并且已经开发了一个(46,47)。O2-PLS通过将数据集中的变化划分为它们之间的联合变化,以及特定于每个数据集且彼此不相关的变化,从而增加了PLS的可解释性(48)。虽然PLS和O2-PLS最初是为化学计量学开发的,但它们最近也被用于组学数据(89,90)。PLS也被扩展到使用内核框架(49),并且开发了一个内核PLS和O2 PLS的组合版本(50)。
与CCA一样,PLS是为两个组学开发的。MBPLS (Multi Block PLS)将模型扩展到两个以上的组学(91),并且sMBPLS增加了稀疏性约束。sMBPLS是专门为组学数据开发的(51)。它寻找与基因表达组的投影有最大相关性的非基因表达组的投影的线性组合。对于多视图数据集,也存在O2PLS的扩展(52)。
在需要高度可解释性的情况下,CCA和PLS都可以使用。不同的u1 k和u2 k向量对是患者之间的相关性(或协方差)最大的向量对。因此,它们可以用于关联来自不同视图的特性。
另一种基于低维协方差最大化的方法是MCIA(53),将共惯性分析扩展到两个以上的组学(92)。其目的是寻找所有组学的预测,使得与全局变异轴的协方差平方和最大:maximum,vα m m=1cov2(Xmum, v)。不同组学的预测可以用来评价不同组学之间的一致性(预测之间的距离反映组学之间的分歧程度)。每个投影都可以用作聚类的表示。
3.非负矩阵分解
非负矩阵分解(NMF)假设数据具有内在的低维非负表示,并且非负矩阵将其投影到观察到的组(93)。因此,它只适用于非负数据。对于单个组,用k表示低维。公式为X≈WH,其中X为n × p观测到的组矩阵,W为n × k, H为k × p。目标函数为||X−WH|| 22,通过使用乘法更新规则交替更新W和H,使其最小化,使每次更新后解保持非负(94)。低维表示W可以使用简单的单组算法聚类。与其他降维方法一样,W和H矩阵可以用来更好地理解每个聚类中每个特征的权重。非负性约束使该权重更易于解释。
有几种方法将该模型推广到多组数据。multimf(54)使用以下概化:将每个组态Xm分解为whm。该模型相当于对每个基因组分别执行NMF。组学之间的集成是通过添加一个约束来完成的,即Wm矩阵接近于“一致”矩阵W*。因此目标函数为:αM m=1||Xm−whm ||2 2 + λαM m=1||Wm−W * ||2 2。Kalayeh等人(55)推广了这种方法,以支持特征和样本相似度的权重。(56)通过进一步要求低维表示W*保持样本之间的相似性来扩展multimf(在原始维度上接近的样本必须在W*上接近)。这种方法结合了因式分解和基于相似性的方法。
联合NMF(57)使用不同的公式,其中样本对所有组学具有相同的低维表示:Xm≈WHm。注意,通过写X = WH,其中X和H是通过矩阵串联得到的,这个模型等价于早期积分。联合NMF不直接用于聚类。相反,数据被简化为一个大维度(k = 200), W和Hm中的高值用于将样本和特征与称为“md模块”的模块相关联。作者将联合NMF应用于卵巢癌患者的miRNA、基因表达和甲基化数据,发现与md模块相关的特征之间的功能富集比单基因组模块中获得的富集更为显著。此外,某些模块的患者与其他患者相比预后有显著差异。就像(56)扩展了multiNMF一样,EquiNMF扩展了Joint NMF,使得原始组学中的相似性在较低的维度上保持不变。(58)将NMF扩展到不同的视图可以包含不同的样本,但基于先验知识约束来自不同视图的某些样本属于同一聚类。最后,PVC(59)执行部分多视图聚类。在此设置中,并非所有示例都必须具有针对所有视图的测量值。
multi - mf和Joint NMF之间的差异类似于前面描述的基于相似性的方法之间的差异。multimf允许不同的组学具有不同的表示,其中它们之间的相似性由参数控制。因此,它可以用于不同组学不期望具有相同低维表示的情况。
4.矩阵tri-factorization三因子分解
(60)中提出的另一种分解方法是三矩阵分解。在这个框架中,每个输入组被视为描述两个实体(即行和列)之间的关系。例如,在具有患者基因表达和DNA甲基化两个组学的数据集中,有三个实体,即患者、基因和CpG位点。基因表达矩阵描述了患者与基因之间的关系,而甲基化矩阵描述了患者与CpG位点之间的关系。
每个描述实体i和j之间关系的维数为ni × nj的组矩阵Rij被分解为Rij = Gi Si jGt j,其中Gi和Gj分别为实体i和j提供了低维表示,属于di维数为ni × ki和nj × kj,其中Sij是一个维数为ki × kj的组特定矩阵。在NMF中,Gi矩阵是非负的。在所有具有实体i的组学中使用相同的Gi矩阵,从而实现数据集成。在上面的例子中,基因表达和DNA甲基化组学都将使用相同的G矩阵来表示患者,但不同的矩阵来表示基因和CpG位点。在该模型中,可以选择性地使用描述基因和CpGs之间关系的附加矩阵。这是矩阵三因子分解的一个主要优点,因为它允许合并不同实体之间先前已知的关系,而无需更改输入组矩阵。(60)对可以鼓励实体具有类似陈述的表述增加了约束。该框架被应用于生物信息学中的各种问题,包括在监督设置中:它被用于执行基因功能预测(60)和患者生存回归(95)。
5.凸配方
大多数基于因子分解的方法的缺点是它们的目标函数不是凸的,因此优化过程不一定达到全局最优,并且高度依赖于初始化。这个问题的一个解决方案是将降维表述为一个凸问题。White等人(61)放宽了CCA的条件,并定义了它的一个凸变体。在降低图像噪声方面进行了性能评估,但该方法也可用于聚类。然而,与CCA一样,该方法只支持两个视图。Guo(62)提出了一种不同的降维凸公式,用于前面提出的一般分解框架,它最小化αM m=1||Xm−BPm||2 F + γ ||B||2,1。||·|| 2,1是l2, 1范数,即矩阵行欧几里得范数之和。因此,这种放松支持多个视图。LRAcluster(16)也使用矩阵分解,并具有凸目标函数。
6.基于张量的方法
多组数据分解方法的一个自然扩展是使用张量,它是高阶矩阵。(63)中提出了一种这样的方法。该方法将每个组矩阵写成Xm = ZmXm + Em, diag(Zm) = 0,其中Zm是一个n × n矩阵,Em是误差矩阵。其思想是,每个组中的每个样本都可以表示为其他样本的线性组合(因此diag(Zm) = 0约束),并且它在该基(Zm)中的表示可以用于聚类。为了整合不同的视图,将不同的Zm矩阵合并为一个三阶张量Z。目标函数鼓励Z是稀疏的,并且Em误差矩阵有一个小范数。
统计方法
统计方法对数据的概率分布进行建模。其中一些方法将样本视为来自不同的集群,其中每个集群定义数据的分布,而其他方法则不显式地使用模型中的集群结构。统计方法的一个优点是,在确定分布函数时,它允许将生物学知识作为模型的一部分。这既可以使用贝叶斯先验,也可以通过选择概率函数来实现。例如基因表达数据采用正态分布。统计框架的另一个优势是它们能够做出“软”的概率决策。例如,统计方法不仅可以将样本分配给集群,还可以确定样本属于该集群的概率。对于大多数公式,参数估计计算困难,并使用不同的启发式。贝叶斯框架下的几种模型允许样本属于不同组学中的不同簇。
1.iCluster和iCluster+
iCluster(15)假设数据来源于低维表示,这决定了每个样本的簇隶属度:Xmt = WmZ + m,其中Z为k x n矩阵,Wm为组特定的pm x k矩阵,k为簇数,m为正态分布噪声矩阵。该模型类似于其他降维模型,但这里噪声的分布是明确的。在该模型下,iCluster通过对稀疏Wm矩阵的额外正则化来最大化观测数据的似然。使用类似em的算法进行优化,随后在数据Z的较低维度表示上运行k-means以获得最终的聚类分配。利用基因表达和拷贝数变异,将聚类应用于乳腺癌和肺癌。最近,在一项癌症分析中,iCluster还被用于对33种癌症的1万多个肿瘤进行聚类(96)。注意,通过将所有Wm矩阵连接到单个W矩阵,并将模型重写为Xt = WZ + , iCluster可以被视为一种早期的集成方法。
iCluster的运行时间随着特征数量的增加而快速增长,因此在使用它之前,特征选择是必不可少的,如(29)所示。Shen等人(15)在分析中只使用位于一条或两条染色体上的基因。
由于iCluster的模型使用矩阵乘法,它需要实值特征。一个名为iCluster+(64)的扩展包括数字、分类和计数数据的不同模型,但保持了数据源自低维矩阵z的想法。对于分类数据,iCluster+假设以下模型:
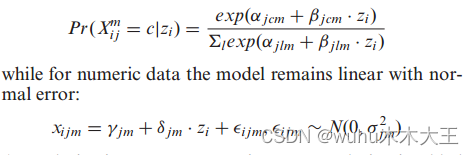
在似然中加入鼓励稀疏解的正则化项,并采用蒙特卡罗牛顿-拉夫森算法进行参数估计。Z矩阵与iCluster中一样用于聚类。基于iCluster+的iCluster的最新扩展是iCluster bayes(65)。该方法将iCluster+中的正则化替换为全贝叶斯正则化。这种替换导致更快的执行,因为算法不再需要为iCluster+的正则化调整参数。
2.PARADIGM
PARADIGM(66)是模拟细胞过程和不同组学之间关系的最明确的方法。对于每个样本和每个细胞路径,创建一个因子图,表示该路径中不同实体的状态。作为一个简并的例子,一条通路可能包括代表该通路中每个基因的mRNA水平的节点,以及代表这些基因拷贝数的节点。因子图中的每个节点可以是激活的、名义的或不激活的,因子图结构定义了这些激活级别上的分布。例如,如果一个基因有高拷贝数,它就更有可能被高度表达。然而,如果该基因的抑制因子高度表达,则该基因更有可能失活。PARADIGM推断未测量的细胞实体的活动,以最大化因子图的可能性,并为每位患者输出每个实体的活动评分。这些分数用于从不同组织中对癌症患者进行分类。
PARADIGM的模型不仅可以用于聚类。例如,PARADIGM-shift(97)通过发现因子图中基于上游实体的预测表达值与使用下游实体的预测表达值不同的基因,来预测功能丧失和功能获得突变。然而,PARADIGM在很大程度上依赖于已知的交互,并且需要对每个组进行特定的建模。它也相当局限于细胞水平;例如,尚不清楚如何将描述每位患者微生物组组成的组学纳入模型。
3.结合特定组和全局聚类
到目前为止讨论的所有方法都假设在不同组学之间存在一致的聚类结构,并且以综合的方式分析聚类将比单独分析每个组学更准确地揭示这种结构。然而,对于生物医学数据集来说,情况并非如此。例如,目前尚不清楚癌症肿瘤的甲基化和表达谱是否真的代表相同的潜在簇结构。相反,有可能每个组学代表了不同的簇结构。有几种方法使用贝叶斯统计来实现这一观点。
Savage等人(67)定义了一个分层Dirichlet过程模型,该模型支持在两个组上聚类。每个样品可以熔接或不熔接。融合样本在两个组学中属于同一簇,而未融合样本在不同组学中可能属于不同簇。融合和未融合样品的模式揭示了两个数据集之间的一致性。该模型在PSDF(68)中得到扩展,包括特征选择。Savage等(67)利用基因表达和ChIPchip数据将该模型应用于基因聚类,而(68)利用表达和拷贝数数据将癌症患者聚类。
在MDI(69)中,每个样本在不同的组学中可以有不同的聚类分配。然而,给出的先验条件是,两个组学之间的关联越强,样本就越有可能属于这两个组学中的同一簇。这种关联强度调整了两个组学之间的先验聚类协议。除了这些先验之外,MDI模型还使用了Dirichlet混合模型,并显式地表示了数据在每个簇和组中的分布。由于样本可能属于不同组学中的不同簇,因此没有由算法驱动的全局聚类解决方案。相反,该算法输出的样本集倾向于属于同一集群。
BCC(70)给出了一个不同的贝叶斯公式。像MDI一样,BCC假设一个Dirichlet混合模型,其中数据来自混合分布。然而,BCC确实假设了一个全局集群解决方案,其中每个样本映射到单个集群。假设一个样本属于一个全局集群,它在每个组中属于该集群的概率很高,但它也可能属于该组中的不同集群。参数估计使用吉布斯抽样(98)。BCC对TCGA乳腺癌的基因表达、DNA甲基化、miRNA表达和RPPA数据进行分析。
像MDI和BCC一样,Clusternomics(71)使用Dirichlet混合模型。集群经济学提出了两种不同的表述。在第一种方法中,每个组都有不同的聚类解,全局聚类被表示为每个组的聚类的笛卡尔积。这种方法不执行多组数据集的集成。在第二种表述中,全球集群被显式映射到特定于组的集群。这样,并非来自不同组学的集群的所有可能组合都被视为全局集群。
4.基于生存的聚类
多组学聚类广泛应用的领域之一是发现疾病亚型。在这种情况下,我们可以预期不同的疾病亚型具有不同的预后,这一标准通常用于评估聚类解决方案。Ahmad和Frohlich(¨72)开发了多组学聚类的贝叶斯模型,在聚类数据时考虑了患者预后。同一簇内的患者特征分布相似,预后也相似。这种方法并非完全没有监督,因为它考虑了患者生存数据,这些数据也用于评估解决方案。Coretto等人(73)也开发了一种考虑生存的概率聚类方法,与(72)相比,它支持大量的特征,而(72)只使用几十个特征。由于生存数据被用作模型的输入,因此与其他方法相比,这种方法给出的集群生存差异更大也就不足为奇了。这在多形性胶质母细胞瘤的数据(72)和几种癌症类型的数据(73)中得到了证实,两者都来自TCGA。
5.深度多视图方法
机器学习的最新发展是深度学习算法的出现(99)。这些算法使用多层神经网络来执行各种计算任务,并被发现在图像识别(100)和文本翻译(101)等多个领域提高了性能。神经网络和深度学习也被证明对多视图应用很有用(102),包括无监督特征学习(37),(103)。学习到的特征可以用于聚类,就像前面描述的DeepCCA一样。深度学习已经广泛用于生物医学数据分析(104)。
最近深度学习在多组学数据中的应用包括(74)和(75)。Chaudhary等人(74)使用自编码器,这是一种深度学习降维方法。作者在RNA-seq、甲基化和miRNA上进行了测试以便聚类肝癌患者。该体系结构实现了一种早期集成方法,将来自不同组学的特性连接起来。自动编码器为每个患者输出一个表示。从这个表示的特征被测试与生存的关联,并使用显著相关的特征来聚类患者。获得的群集存活率有显著差异。该结果与使用原始特征的类似分析以及使用PCA(主成分分析)而不是自动编码器学习的特征进行比较。然而,这项工作中的分析并不是无监督的,因为特征选择是基于患者生存的。
Liang等人(75)采用了不同的方法。他们使用深度信念网络(105)分析卵巢癌的表达、甲基化和miRNA数据,该网络明确考虑了数据的多组学结构。该体系结构包含独立的隐藏层,每个隐藏层都有来自一个组的输入,随后的层接收来自所有单组隐藏层的输入,从而集成了不同的组。学习每个患者在{0,1}上的3D表示,将患者划分为8个簇。聚类结果与所有组学串联的k-means聚类结果进行了比较,但没有与其他多组学聚类方法进行比较。
深度学习算法通常需要很多样本和很少的特征。它们使用大量的参数,这使得它们容易过度拟合。当前的多基因组数据集具有相反的特征——它们具有许多特征,并且至少少了一个数量级的样本。这里展示的作品在他们的架构中只使用了几层来克服这一限制,相比之下,最先进的成像数据集架构使用了数十层。随着生物医学样本数量的增加,深度多视图学习算法可能对生物医学数据集更有益。
BENCHMARK
为了测试多组学聚类方法的性能,我们比较了TCGA提供的10种癌症类型的9种算法。我们还比较了算法在组成多组数据集的每个单组数据集上的性能,以获得适用于单组数据的算法。选择这9种算法来代表不同的多组学聚类方法。在每种方法中,我们都选择了具有可用软件和明确使用指南的方法(例如,我们选择pin而不是COCA作为后期集成方法,因为COCA没有明确说明每个单个基因组应该如何聚类),并且这些方法被广泛使用,因此这些方法的比较将对社区最有帮助。三种算法是早期的集成方法:LRAcluster,以及k-means和谱聚类,将组学连接到单个矩阵中。对于基于相似性的算法,我们使用SNF和rMKL-LPP。对于降维,我们使用MCCA(39)和multimf。我们选择iClusterBayes作为统计方法,而pin作为后期集成方法。
这十个数据集包含癌症肿瘤多组学数据,其中每个数据集都是不同的癌症类型。所有的数据集包含三个组学:基因表达、DNA甲基化和miRNA表达。患者数量从AML的170例到BIC的621例不等。关于数据集和癌症类型缩略语的详细信息见补充文件2。
为了评估集群解决方案的性能,我们使用了三个指标。首先,我们使用logrank检验(106)来测量所获得的群集之间的生存差异。使用该测试作为度量标准,假设如果患者群的生存率显著不同,那么它们在生物学意义上是不同的。其次,我们测试了群集中临床标签的富集程度。我们选择了六个临床标签进行富集测试:性别、诊断时年龄、病理T、病理M、病理N和病理分期。后四个参数是离散的病理参数,测量肿瘤的进展(T),转移(M)和淋巴结癌(N),以及总进展(病理分期)。离散参数的富集计算使用2独立性检验,数值参数使用KruskalWallis检验。并非所有的临床参数都适用于所有的癌症类型,因此总共有41个临床参数可用于测试。最后,我们记录了每个方法的运行时间。我们在评估聚类质量时没有考虑计算度量,如异质性、同质性或剪影分数(107),因为不同的方法对特征执行不同的归一化(有些甚至执行特征选择)。关于存活和表型数据的详细信息见补充文件2。
为了推导logrank检验、2独立性检验和Kruskal-Wallis检验的p值,假设这三个检验的统计量具有2分布。然而,对于logrank测试和2测试,这种近似对于小样本量和不平衡的簇大小是不准确的,特别是对于较大的测试统计量(例如在(108)中,对于两个簇的logrank测试)。因此,我们在这里报告的p值是使用排列检验来估计的(即,我们在样本之间排列聚类标签并使用检验统计量来获得经验p值)。我们确实观察到基于置换检验和基于近似的p值之间存在很大差异,对于logrank检验和临床参数的富集。关于排列测试的更多细节见补充文件1。排列检验后,临床标签的p值对多个假设进行校正(因为测试了几个标签),对每种癌症类型和方法使用Bonferroni校正,显著性水平为0.05。统计分析结果见补充文件3。
我们将所有9种方法应用于10个多组学数据集,以及包含它们的30个单组学矩阵。唯一的例外是MCCA,我们不能应用于单基因组数据,以及pin,它在所有BIC数据集上始终崩溃*。所有方法都在Windows机器上运行了,除了iCluster,它在Linux集群上并行使用多达15个节点。一般来说,我们根据作者的建议选择方法的参数。如果作者建议进行参数搜索,则进行参数搜索,并根据作者建议选择最佳解决方案,而不考虑用于评估的生存和临床参数。我们为这些方法报告的运行时包括参数搜索。其基本原理是,基准测试旨在记录用户在结果质量和总运行时间方面如何运行这些方法。有关硬件、数据预处理和方法应用的详细信息见补充文件1。完整的聚类结果见补充文件4。所有处理过的原始数据都可以在http://acgt.cs.tau.ac上获得。使用的所有软件脚本都可以在https://github.com/Shamir-Lab/ multi - omics - cancer - benchmark/上获得。
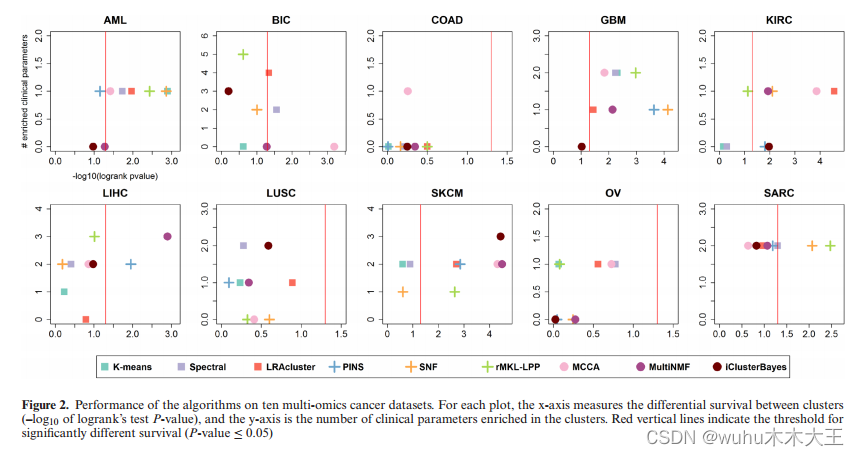
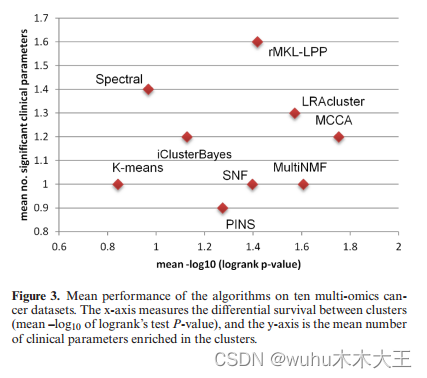
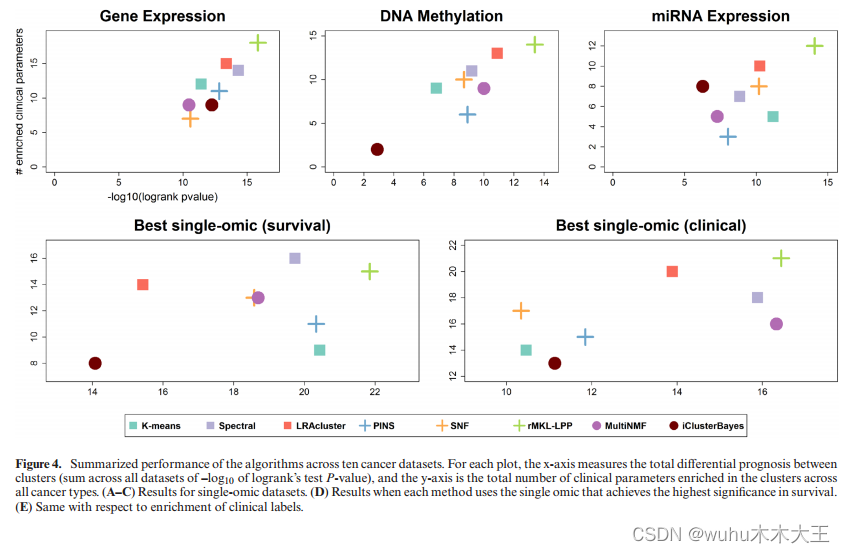
图2描述了基准方法在不同癌症数据集上的性能,图3和图4分别总结了所有癌症类型的多组学数据和每个单组学的性能。在差异生存或丰富的临床参数方面,没有任何算法始终优于所有其他算法。在生存方面,MCCA具有总最佳预后值(-log10 p值的总和= 17.53),而multimf排名第二(16.07),LRACluster排名第三(15.72)。由于异常值的存在,p值的总和可能存在偏差,因此我们还计算了一种方法的解决方案获得显著不同存活率的数据集的数量。这些结果报告在表2中。在这里,除了iClusterBayes之外,所有针对多组学或多视图数据开发的方法都至少有四种癌症类型,其生存率存在显着差异。MCCA和LRACluster有5个。这些癌症类型对所有算法来说都是不一样的。

rMKL-LPP获得的重要临床参数总数最多,共有16个参数。光谱聚类排名第二,有14个,LRAcluster有13个。在存活方面效果较好的MCCA和MultiNMF分别只有12个和10个富集参数。对于所有癌症类型,rMKL-LPP并没有优于所有其他方法。例如,它有一个SKCM的丰富参数,而其他几个方法有两个或三个。我们还考虑了算法至少具有一个丰富临床标签的癌症类型的数量(表2)。rMKL-LPP、光谱聚类、LRACluster和MCCA在8种癌症类型中都有富集,而MCCA总共只有12个富集参数。总体而言,rMKL-LPP在生存和临床富集方面优于MCCA, LRACluster和multiNMF之外的所有方法。MCCA、LRACluster和multimf具有较好的预后价值,但临床标签的富集程度较低。
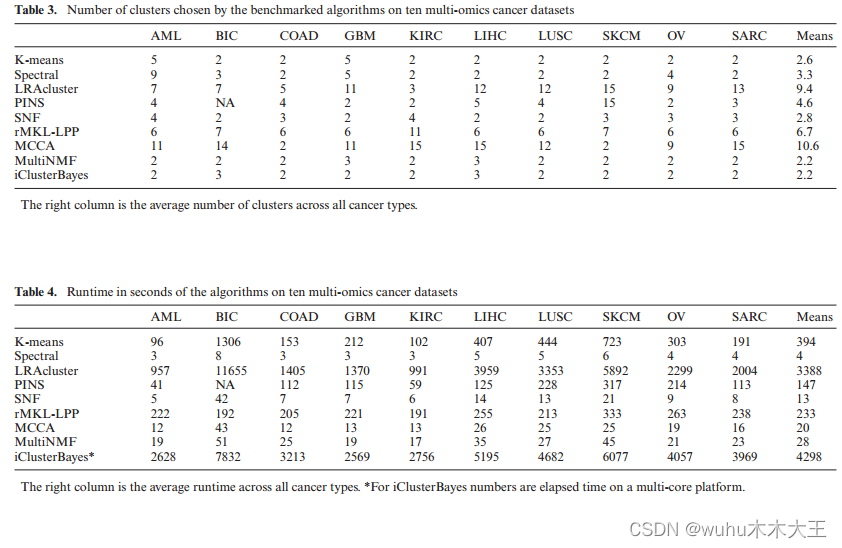
每种方法确定每个数据集的簇数。表3列出了这些数字。这些数字在不同的方法之间差别很大,从2到3个(iCluster和MultiNMF)到平均超过10个(MCCA)。MCCA、LRACluster和rMKL-LPP将数据划分为数量相对较多的聚类(平均分别为10.6、9.4和6.7),并且表现良好,这可能说明将癌症患者划分为更多的聚类可以提高预后价值和临床意义。在logrank和临床富集试验中,通过对其卡方统计量具有更大的自由度来控制更高的簇数。
表4中报告了不同方法的运行时。请注意,如前所述,在集群上运行了iClusterBayes,而其他方法则在台式计算机上运行。除了LRAcluster和iCluster之外,所有方法对每个数据集的平均处理时间都不到10分钟。LRAcluster和iClusterBayes在每个数据集上分别花费了56分钟和72分钟。
图4还显示了针对单组数据的基准测试方法的性能。虽然有几种方法在单组数据集上的性能较差,但有些方法的性能更好。例如,基因表达的rMKL-LPP在单组学和多组学数据集中都获得了最多的临床参数(18)。基因表达溶液也比多组学溶液具有更好的预后价值。
为了进一步测试单基因组数据集的分析与多基因组数据集的比较,我们为每个数据集和方法选择了能够提供最佳生存和临床富集结果的单基因组。在本分析中,rMKL-LPP具有最高的临床参数富集总数(21)和最高的总生存意义(21.86)。单组学数据集的运行时间、簇数、生存期和临床富集分析见补充文件1和3。这些结果表明,与单独分析单组学数据相比,多组学数据分析并不总是提供更好的预后价值和临床意义,特别是当每种癌症类型使用不同的单组学时。
讨论
本文综述了多组学和多视图聚类的研究方法。在我们对10个癌症数据集的测试中,总体而言,rMKL-LPP在临床富集方面表现最好,并且在生存方面优于除MCCA和MultiNMF之外的所有方法。MCCA和multiNMF的高性能是值得注意的,因为这些是多视图方法,不是专门为组学数据开发的(尽管MCCA被应用于它)。
在这篇综述中,我们提供了关于不同方法和算法的优缺点的指南。在基准测试中,没有一种方法在任何评估标准上始终优于所有其他方法。虽然有些方法表现得很好,但我们不能由此得出结论,认为它们应该永远是首选。我们也无法确定一个“最佳”的整合方法,但有趣的是,在生存方面表现最好的两个方法是降维方法。
在将多视图聚类方法应用于多组数据时,应该仔细考虑,因为这些数据具有多视图方法不一定考虑的特征。这些特征中最突出的是相对于样本数量的大量特征。例如,CCA对每个组的协方差矩阵进行反转。当特征多于样本时,该矩阵是不可逆的,需要进行稀疏正则化。多组数据的另一个特征是不同组中特征之间的依赖关系,但在给定聚类结构的情况下,一些多视图算法假设组具有条件独立性。这种依赖很少被考虑,因为它极大地增加了模型的复杂性。当前基因组数据类型的另一个特征是,由于细胞调节,它们具有内在的低维表示。许多方法都利用了这一特性。
在我们的基准测试中,单组学数据有时比多组学数据给出更好的结果。当每种算法都选择了每种癌症类型的“最佳”单组时,这种情况得到了加强。这些结果质疑当前普遍存在的多组学分析和多组学聚类的假设。
几种方法可以改善多组学分析的结果。首先,本文开发并回顾了在不同组学中提出不同聚类的方法,但没有包括在基准中,因为尚不清楚如何比较不输出全局聚类解决方案的算法与输出全局聚类解决方案的算法。这些方法可能对只出现在某些组学中的强烈信号更敏感。其次,未来的算法可以以与当前算法相同的方式执行组选择。在基准测试中,我们让每种方法根据分析结果为每种癌症类型选择一个单组学,而这些分析结果通常无法用于实际数据。过滤具有矛盾信号的组学的方法可能获得更清晰的聚类。最后,虽然一些多组学聚类方法包含了先前的生物学知识,但很少有方法包含了组学之间或不同组学特征之间的关系知识。几种统计方法包括某种形式的描述生物建模MDI基于组学的相似性来调整不同组学中聚类方案的相似性。然而,这些方法不能模拟组学之间的生物学关系。一个明显的例外是PARADIGM,它表述了不同组学之间的关系。然而,它也需要对相互作用的生化后果有准确的先验知识,而这些知识通常是不可用的。建模组学之间关系的方法可能受益于额外的生物学知识,即使没有建模整个途径。例如,我们可以在模型中加入启动子甲基化与基因表达反相关的事实。据我们所知,这些方法仅用于拷贝数变异和基因表达数据(如:(109)),而不是在集群的背景下。
在我们使用的统计检验中,我们发现从卡方近似得到的p值与从排列检验得到的p值之间存在很大差异。由于小样本量、小簇大小(在具有大量簇的溶液中)以及logrank测试的低事件数(高存活率),差异特别大。单组和多组方法使用这些p值来评估它们的性能,而logrank p值通常是算法优点的主要论据。p值之间的巨大差异质疑了基于卡方近似的分析的有效性,至少对于TCGA数据是如此。未来的工作必须在数据集中使用精确的或基于排列的Pvalue计算,这些数据集具有与基准测试中使用的数据集相似的特征。
我们执行的基准测试并非没有限制。使用患者生存期来衡量表现,在某种程度上偏向于已知的癌症亚型,这可能已用于治疗决策。此外,生物学上不同的癌症亚型可能有相似的生存期。对于丰富的临床参数也是如此,尽管我们试图选择不会导致这种偏差的参数。然而,这些措施被广泛用于聚类评估,包括在描述一些基准方法的论文中。基准测试的另一个限制是它只检查聚类,而一些方法有额外的目标和输出。例如,在降维算法中,低维数据可以用于分析特征,而不仅仅是患者,例如通过计算几个组学共有的变异轴。在特征分析方面,我们没有测试过的多组算法比单组算法更有优势。最后,尽管我们根据作者给出的指导原则选择了每种基准方法的参数,但明智地微调参数可能会改善结果。

















 7289
7289

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









