







GUIDANCE Server - a web server for assessing alignment confidence score (evolseq.net)
anzaika/guidance: A stable place for guidance source code http://guidance.tau.ac.il/ver2/source.php (github.com)
注意
由于原作者所在地战争原因,目前源代码出现问题,所以请下载上面的github链接。下列出现的guidance.v2.02全部替换为guidance-master即可。
概述
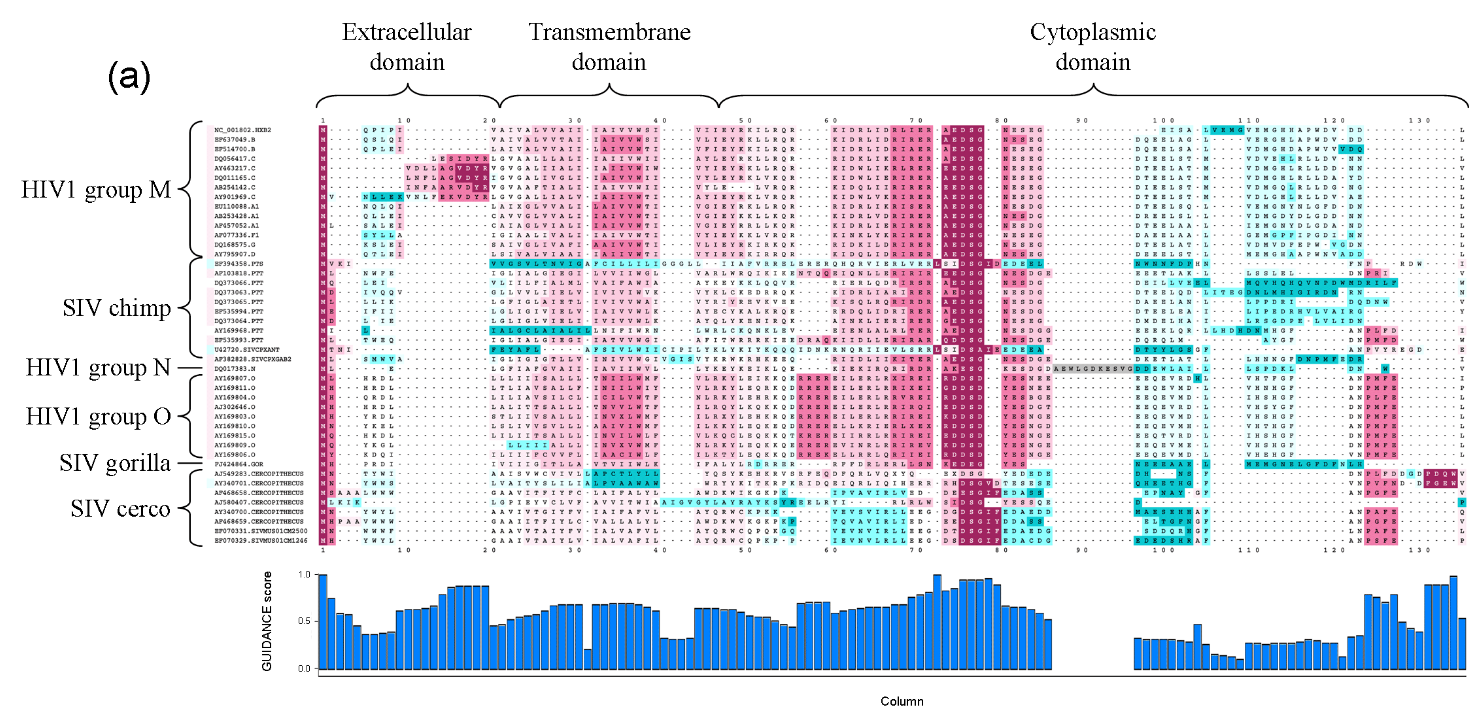
这个软件的作用是调用多序列比对软件,例如MAFFT,MUSCLE等进行比对,并对获得的位点按照系统发育置信度进行打分+可视化。

软件安装
下载,之后执行
tar -xzvf guidance.v2.02.tar.gz
cd guidance.v2.02
make
检查必备软件:
任选其一安装:
-
MAFFT: Type “mafft” and check that you have version 6.712 or newer.
-
Else download and install MAFFT from:
http://mafft.cbrc.jp/alignment/software/ -
PRANK: Type “prank” and check that you have version v.100223 or newer.
-
Else download and install PRANK from:
http://www.ebi.ac.uk/goldman-srv/prank/prank/ -
CLUSTALW: Type “clustalw” and check that you have it insalled
- Else download and install CLUSTALW from:
http://www.ebi.ac.uk/Tools/clustalw2/index.html
- Else download and install CLUSTALW from:
检查必备环境:
GUIDANCE also uses Perl, BioPerl and Ruby:
-
Type “perl -v” and check that you Perl installed.
-
Else download and install it from:
http://www.perl.org/ -
Type “perl -e ‘use Bio::SeqIO’” to check that you have BioPerl.
-
Else download and install it from:
http://www.bioperl.org/ -
Type “ruby --version” to check that you have ruby.
-
Else download and install it from:
http://www.ruby-lang.org/en/
运行
guidance/www/Guidance/guidance.pl
用法:
perl <guidance directory>/www/Guidance/guidance.pl \
--seqFile SEQFILE \
--msaProgram [MAFFT|PRANK|CLUSTALW|MUSCLE] \
--seqType [aa|nuc|codon] \
--outDir FULL_PATH_OUTDIR
必需的参数:
--seqFile:输入的序列文件,需为FASTA格式--msaProgram:使用的多序列比对(MSA)程序--seqType:对齐的序列类型(氨基酸、核苷酸或密码子)--outDir:将自动创建并包含所有输出文件的输出目录 [请提供完整路径而非相对路径]
可选参数:
--program[GUIDANCE2|GUIDANCE|HoT] 默认=GUIDANCE2--bootstraps:引导迭代次数(仅限于 GUIDANCE)。默认值=100。越多越慢。--genCode:遗传密码标识符(仅限于密码子序列)。默认值=1
1) Nuclear Standard
15) Nuclear Blepharisma
6) Nuclear Ciliate
10) Nuclear Euplotid
2) Mitochondria Vertebrate 线粒体脊椎动物
5) Mitochondria Invertebrate
3) Mitochondria Yeast
13) Mitochondria Ascidian
9) Mitochondria Echinoderm
14) Mitochondria Flatworm
4) Mitochondria Protozoan
--outOrder默认=aligned。也可以选择as_input--msaFile:输入的比对文件 - 不推荐,参见概述部分--seqCutoff:置信度截止值,介于0到1之间。默认值=0.6--colCutoff:置信度截止值,介于0到1之间。默认值=0.93--mafft:mafft可执行文件的路径。默认值=mafft--prank:prank可执行文件的路径。默认值=prank--muscle:muscle可执行文件的路径。默认值=muscle--pagan:pagan可执行文件的路径。默认值=pagan--ruby:ruby可执行文件的路径。默认值=ruby--dataset:数据集的唯一名称 - 将用作输出的前缀(默认值=MSA)--MSA_Param:为比对程序传递参数,例如 PRANK 的-F。要传递包含‘-’的参数,每个‘-’前都要加上反斜杠,例如\-F--proc_num:使用的处理器数量(默认值=1)
示例:
guidance \
--seqFile /mnt/e/Scientifc_software/guidance-master/guidance-master/Tem/Tem.fasta \
--msaProgram muscle \
--seqType nuc \
--genCode 2 \
--outDir /mnt/e/Scientifc_software/guidance-master/guidance-master/Tem/outcome \
--bootstraps 5 \
--proc_num 24 \
--dataset TEM \
# 将使用MAFFT比对“10K_1069_hap.fasta”中的序列,并将所有结果输出到目录“/somewhere/protein.guidance”
perl <guidance directory>/www/Guidance/guidance.pl \
--seqFile codingSeq.fas \
--msaProgram PRANK \
--seqType codon \
--outDir /somewhere/codingSeq.guidance \
--genCode 2 \
--bootstraps 30
# 将使用PRANK比对“codingSeq.fas”中的密码子序列,并使用脊椎动物线粒体遗传密码进行翻译,将所有结果输出到目录“/somewhere/codingSeq.guidance”。只进行30次引导迭代,而不是默认的100次(将运行时间缩短3倍)















 1514
1514

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









