







可有可无的摘要
还在为导师布置的刁钻任务发愁?
还在为自己的蛋白质找不到对象(配体)焦虑?
还在为...好吧编不下去了
本文旨在帮助大多数在搜索引擎中苦寻分子对接教程,却因难度过高被劝退的科研小白(是的,就是你),本教程简洁无废话(废话)。全程截图干货,跟着本教程一步步操作,保证你做出好看的分子对接图片,给你导师一份满意的答卷,做不出来你打我!
以下是目录:
目录
话不多说直接进入教程
软件安装
#autodock安装
#MGLTools安装(安装前需要创建文件夹)
#Anaconda安装(安装前需要创建文件夹)
Download Anaconda Distribution | Anaconda
#pymol开源版安装
#需要在anaconda命令行中安装
conda install conda-forge::pymol-open-source
#openbabel的安装
conda install openbabel -c conda-forge
配置工作目录
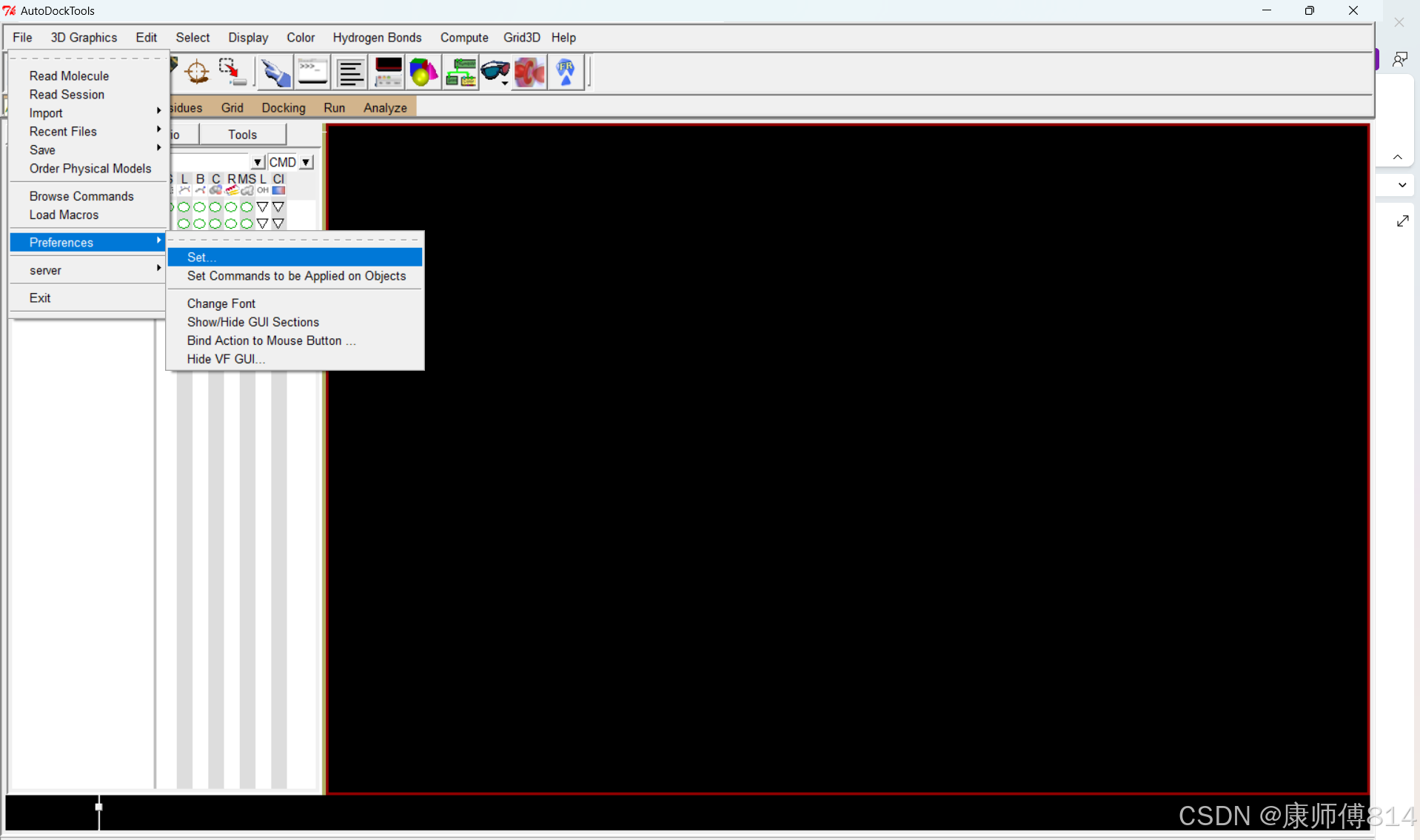
#将Autodock4.2.6安装目录下的autodock4.exe、autogrid4.exe以及mgltools安装目录下的adt.bat复制到一个新的目录下,新目录命名为Autodock(这里我的工作目录为D:\00workdata\Autodock)(工作目录路径中不要有英文
#打开autodocktools,设置工作目录路径
文件准备
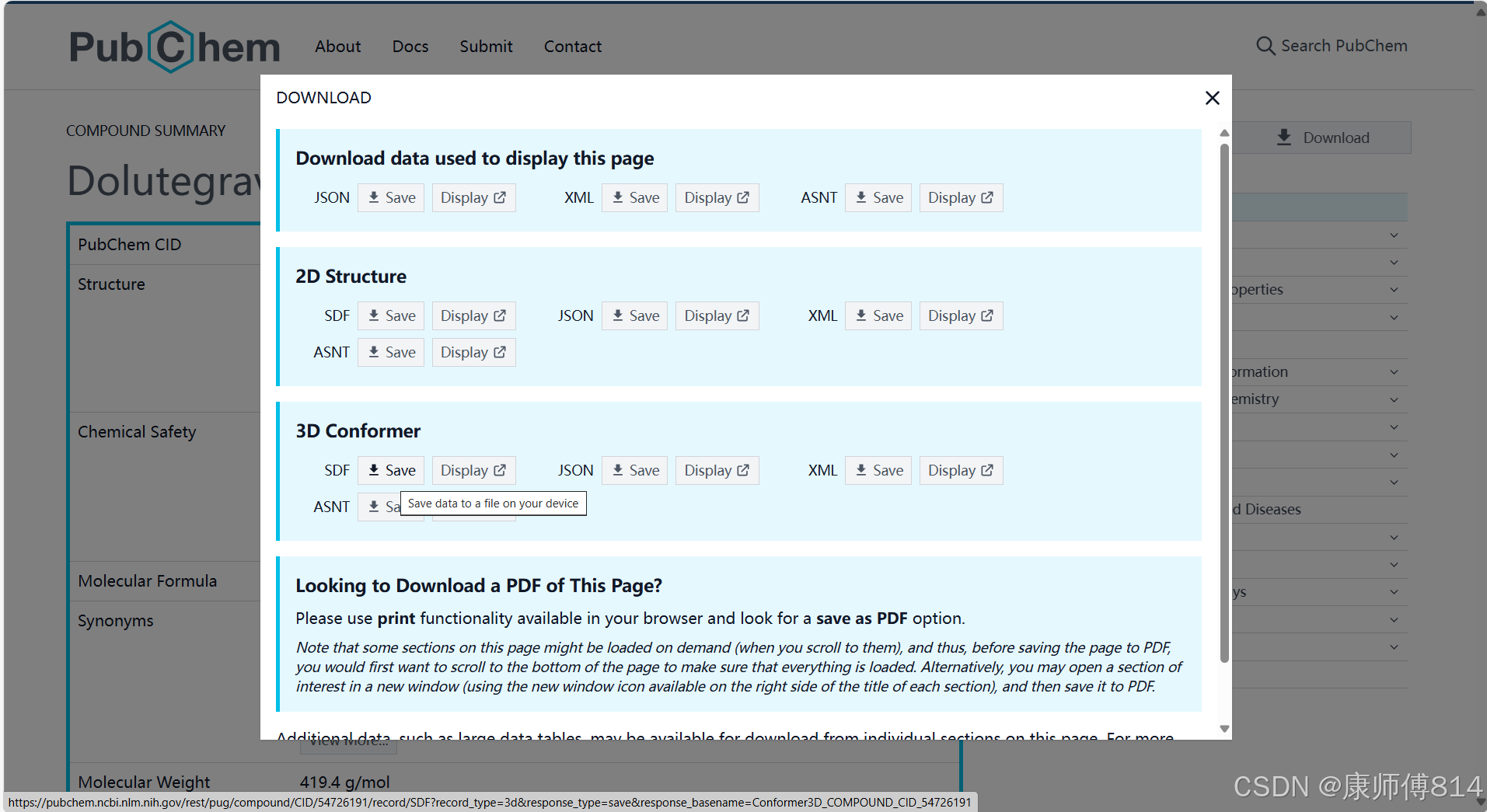
pubchem数据库下载小分子结构SDF文件
PDB数据库下载蛋白质结构
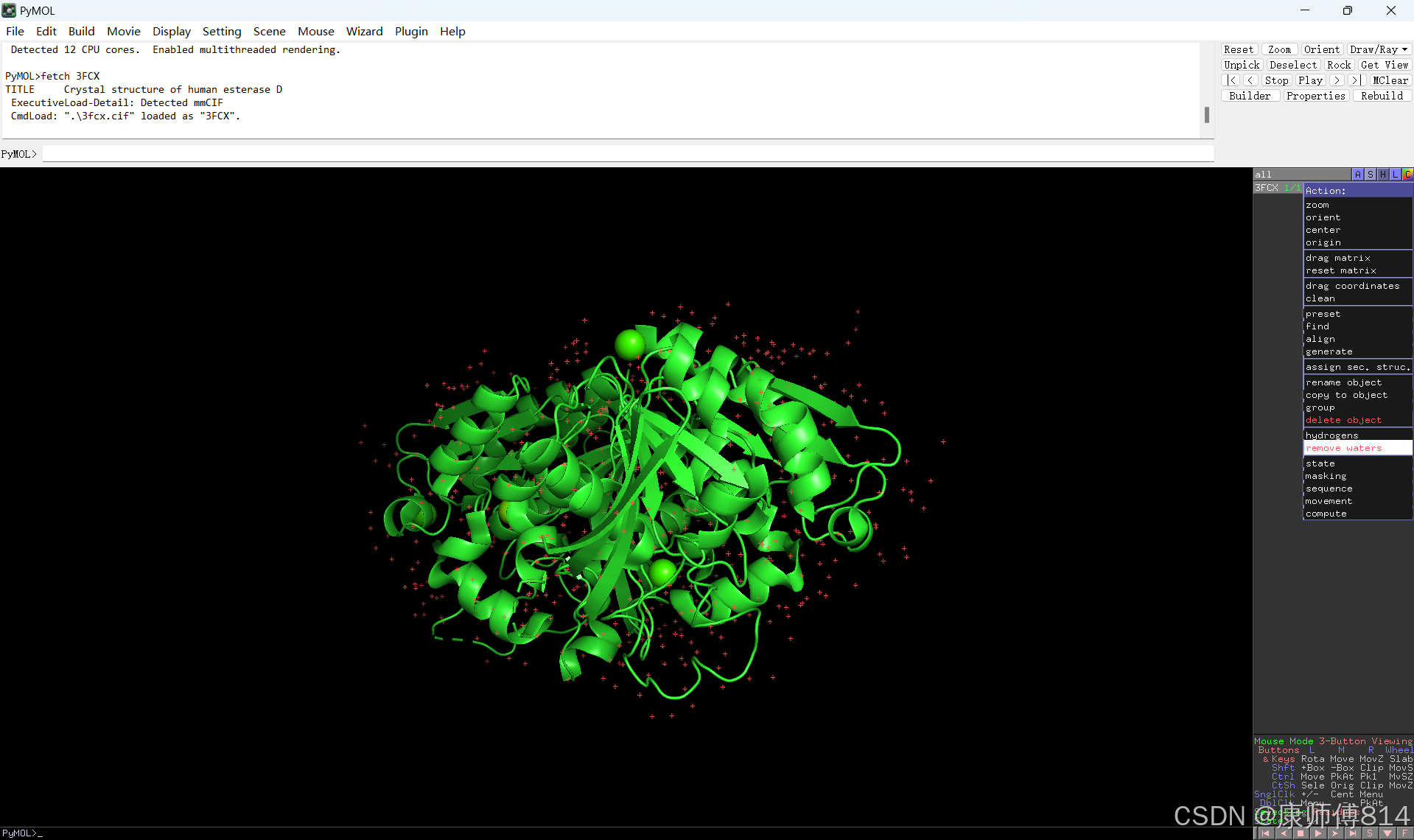
#在PDB网站找到所需的蛋白编号(例如酯酶D为3FCX)
#打开pymol,输入fetch 3FCX,按回车自动下载蛋白3D结构。
#去除水分子,如下图所示,点蛋白编号前面的A,选择去除水分子。点击左上角File-Export Molecule,导出生成PDB文件。命名为ESD.pdb
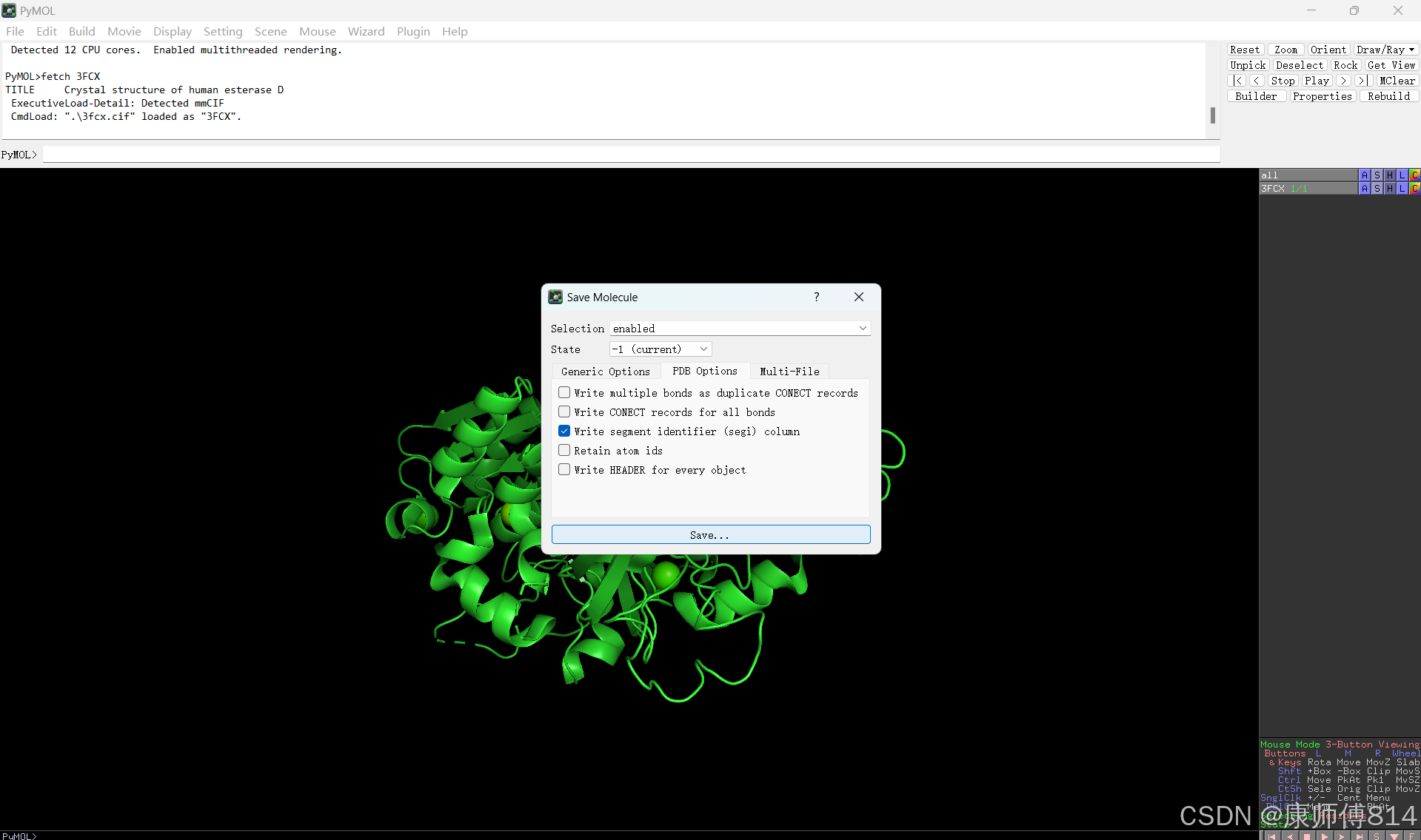
蛋白质文件前处理
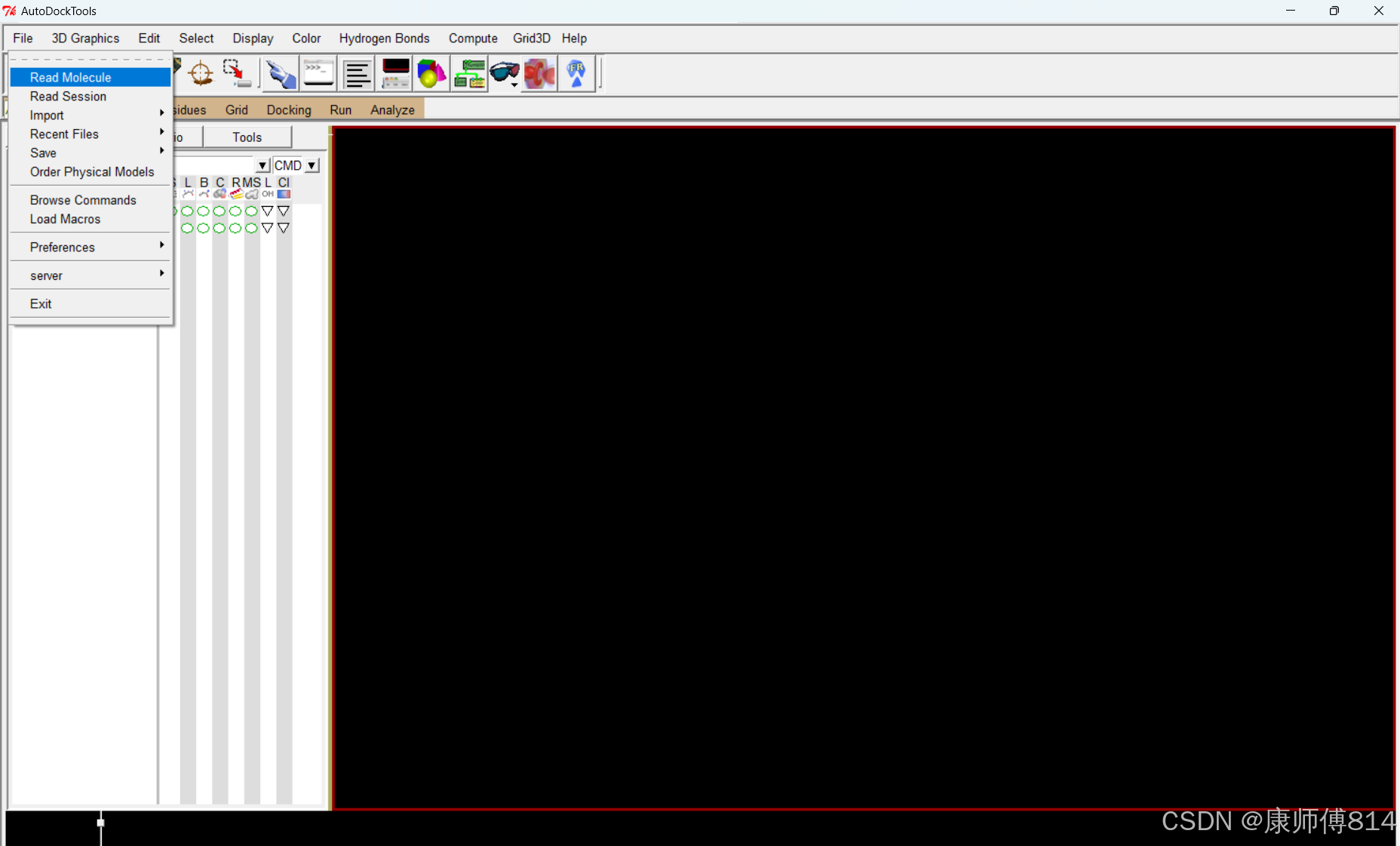
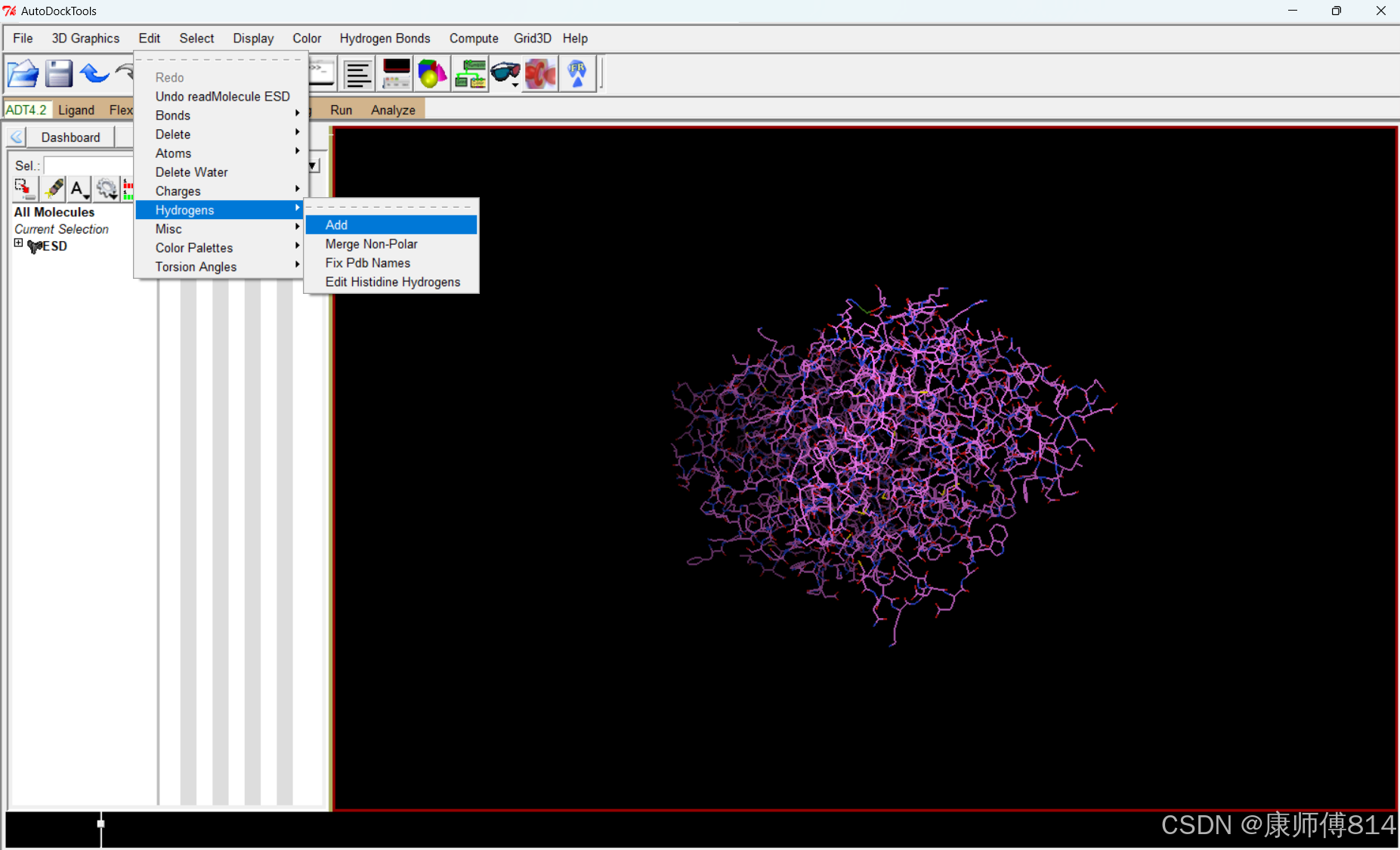
#蛋白质结构加氢
#autodocktools打开蛋白质pdb文件,点击Edit-Hydrogens-Add
#将蛋白质选为受体
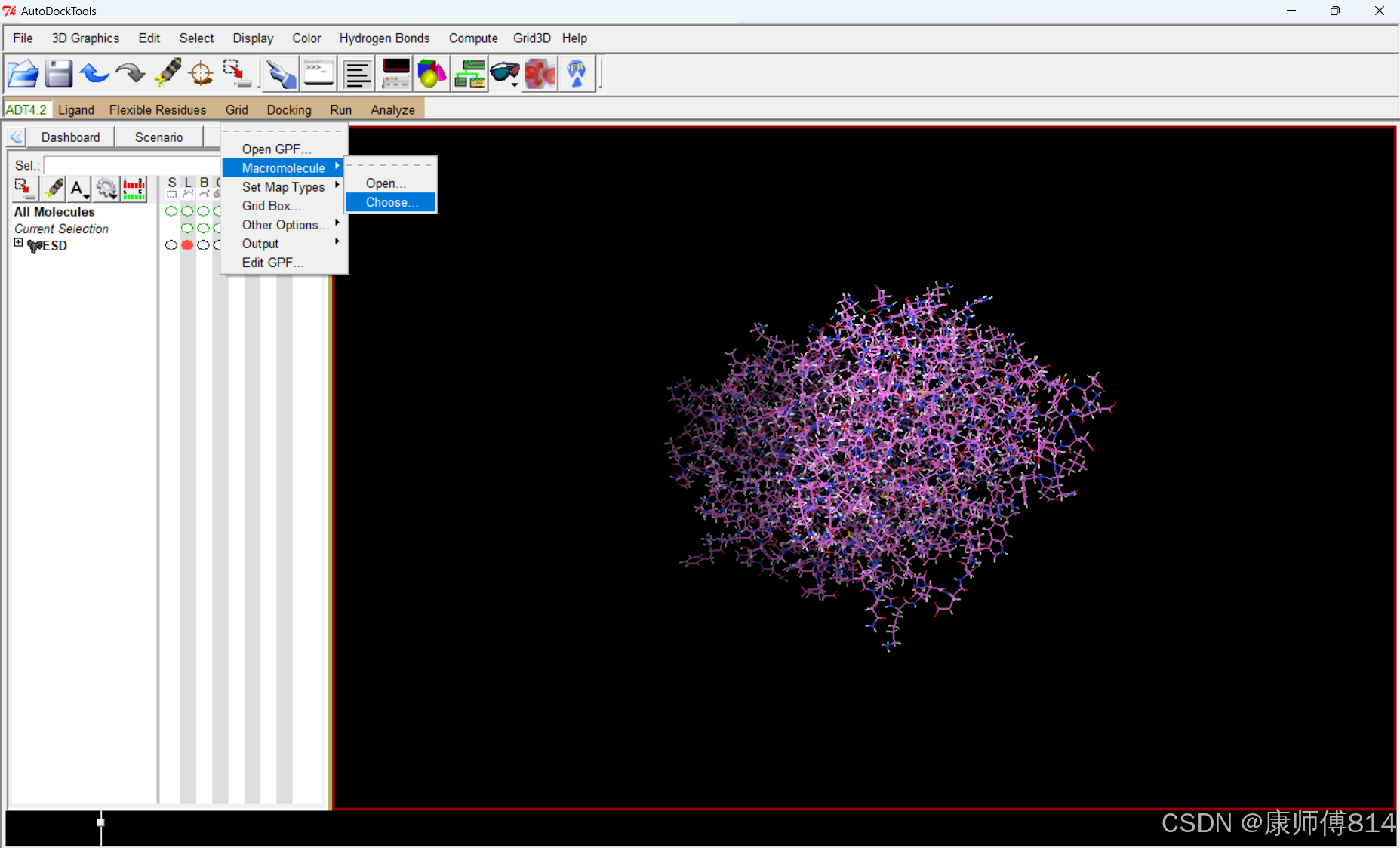
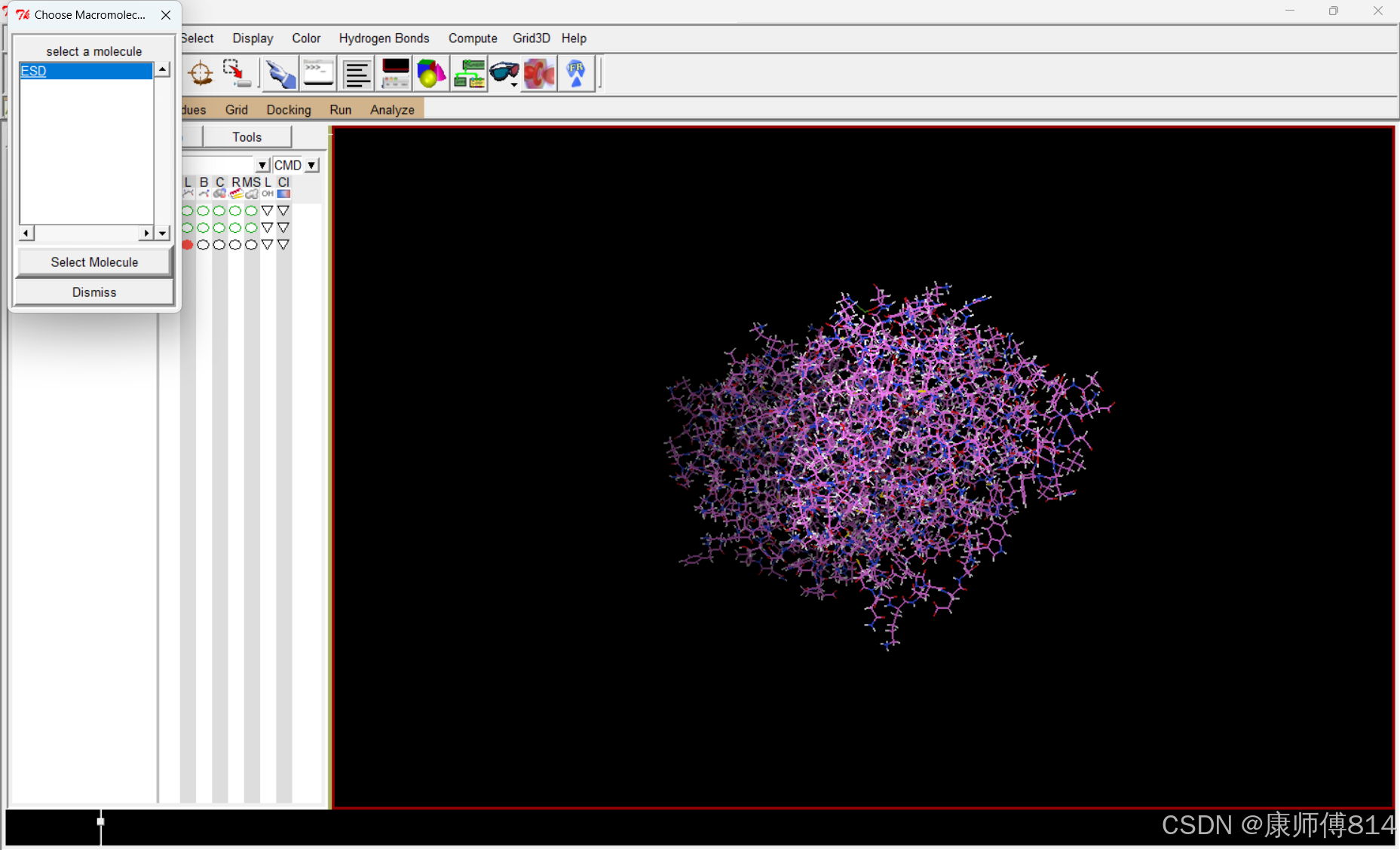
#点击Grid-Macromolecule-Choose,点击蛋白质名称,点击蛋白质名称,点击Select Molecule,根据提示操作保存为ESD.pdbqt
小分子文件前处理
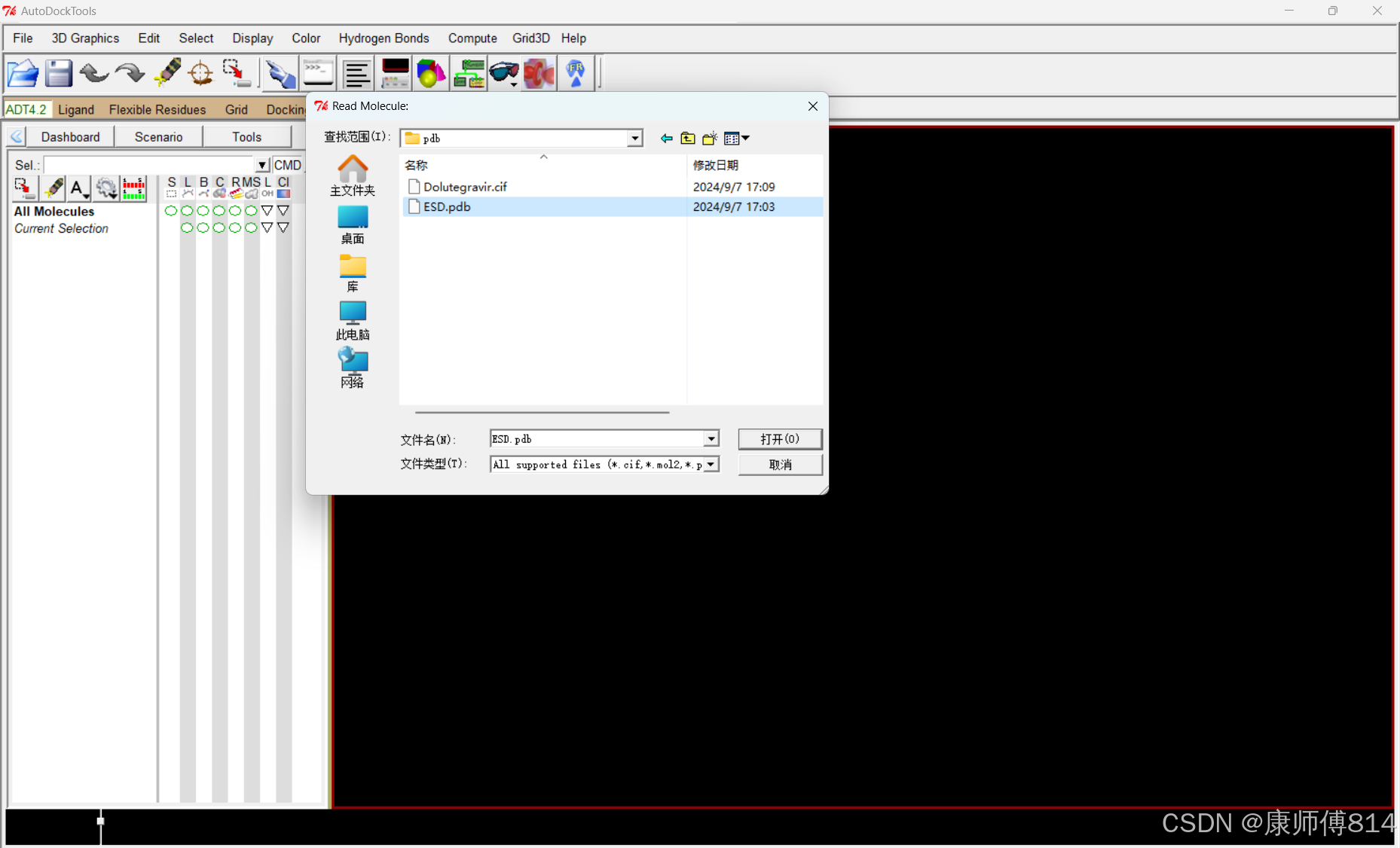
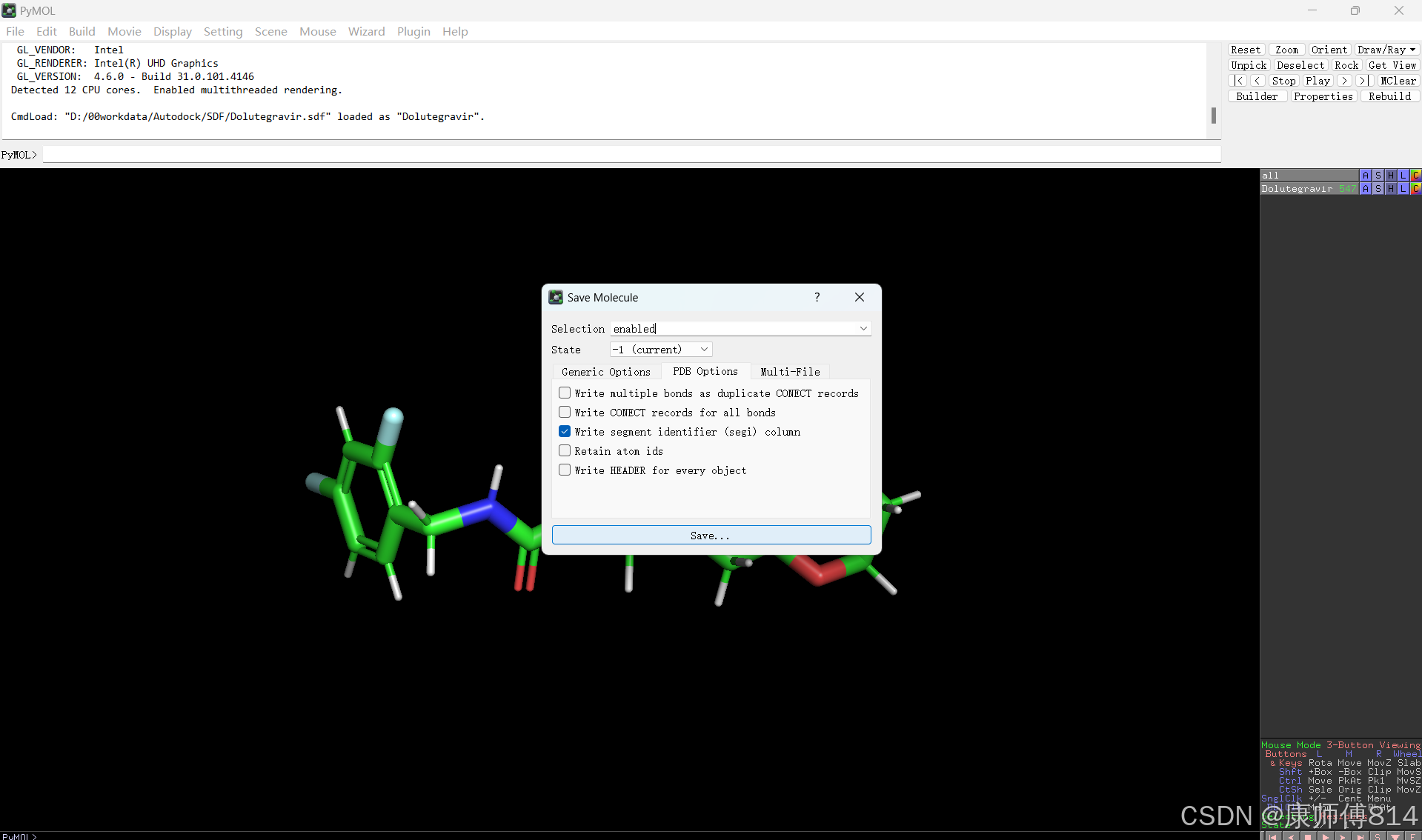
#pymol打开小分子sdf文件,导出为pdb格式
#小分子文件加氢
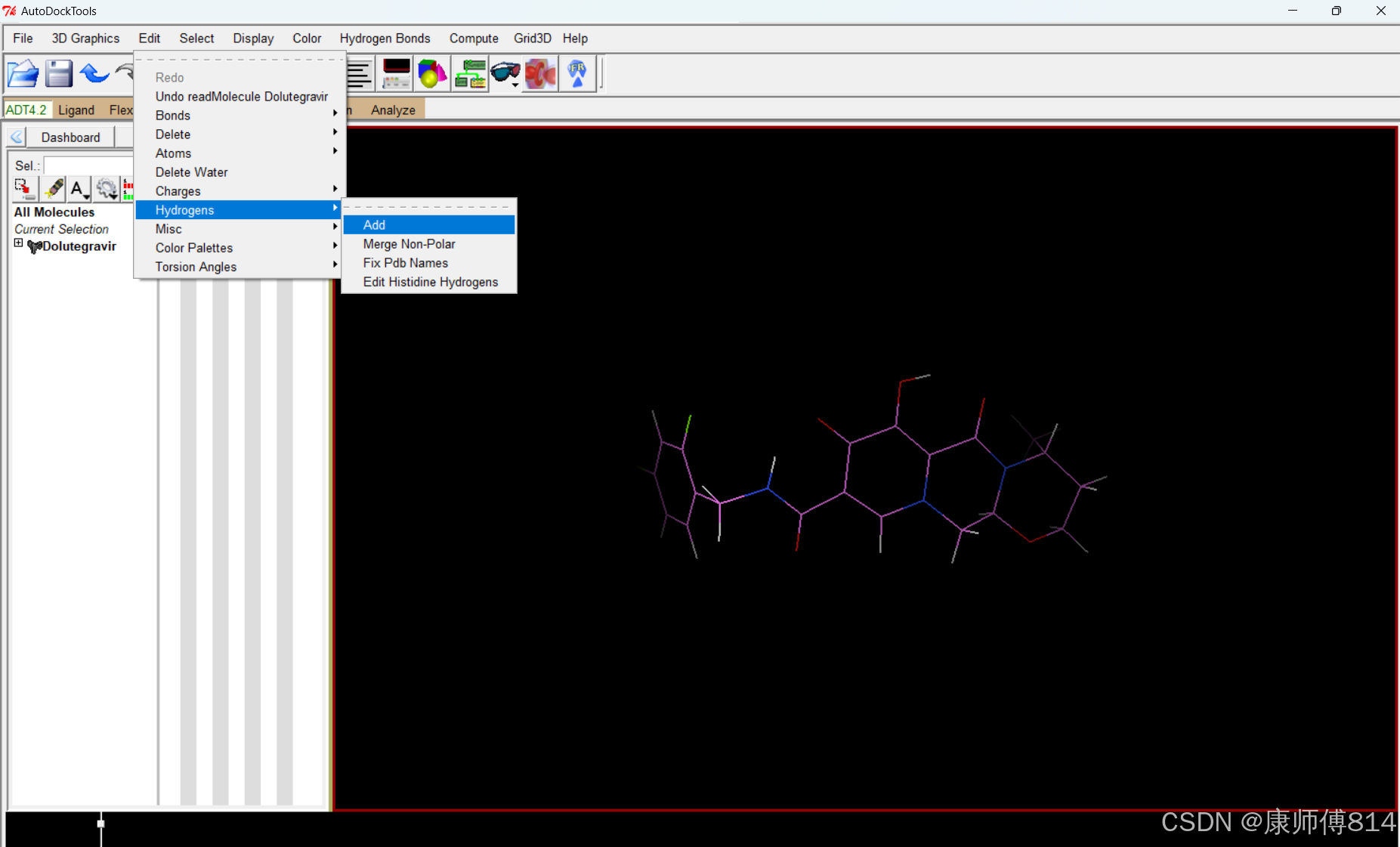
#autodocktools打开小分子pdb文件,点击Edit-Hydrogens-Add
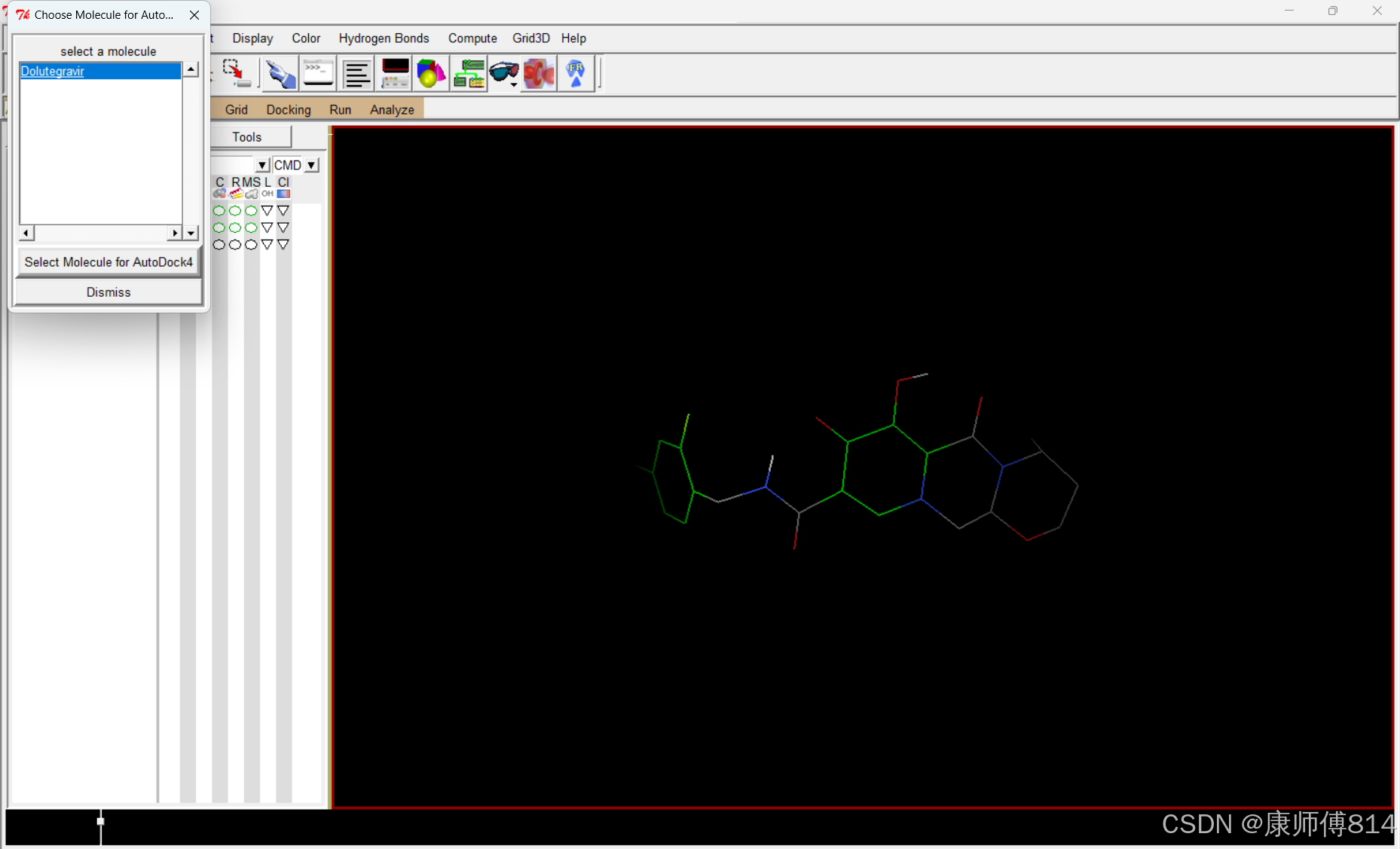
#选择小分子为配体
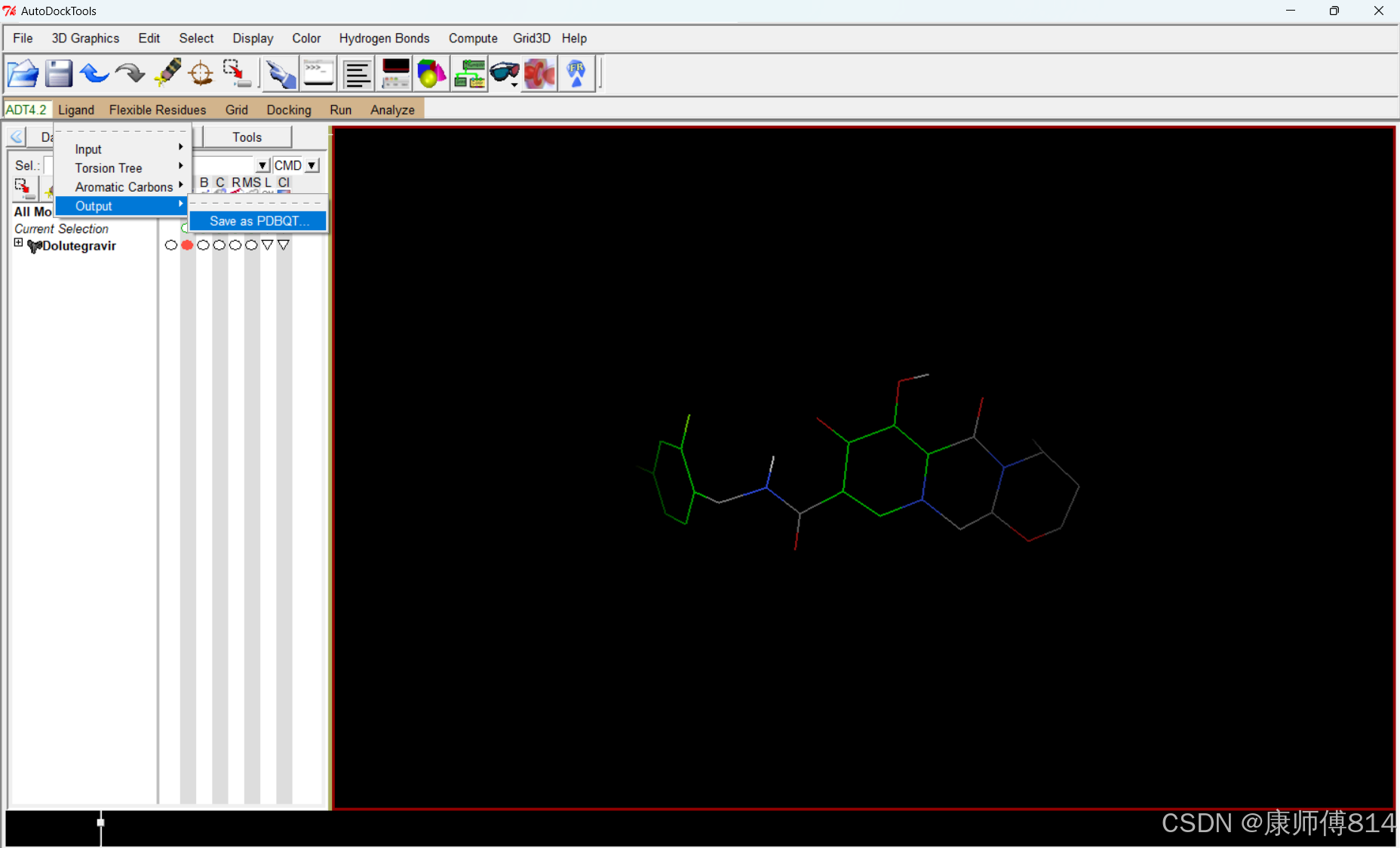
#点击Ligand-Input-Choose,点击小分子名称,点击Select Molecule for Autodock4,点击Ligand-Output-Save as PDBQT,保存为pdbqt文件

进行autogrid
导入配体和受体文件
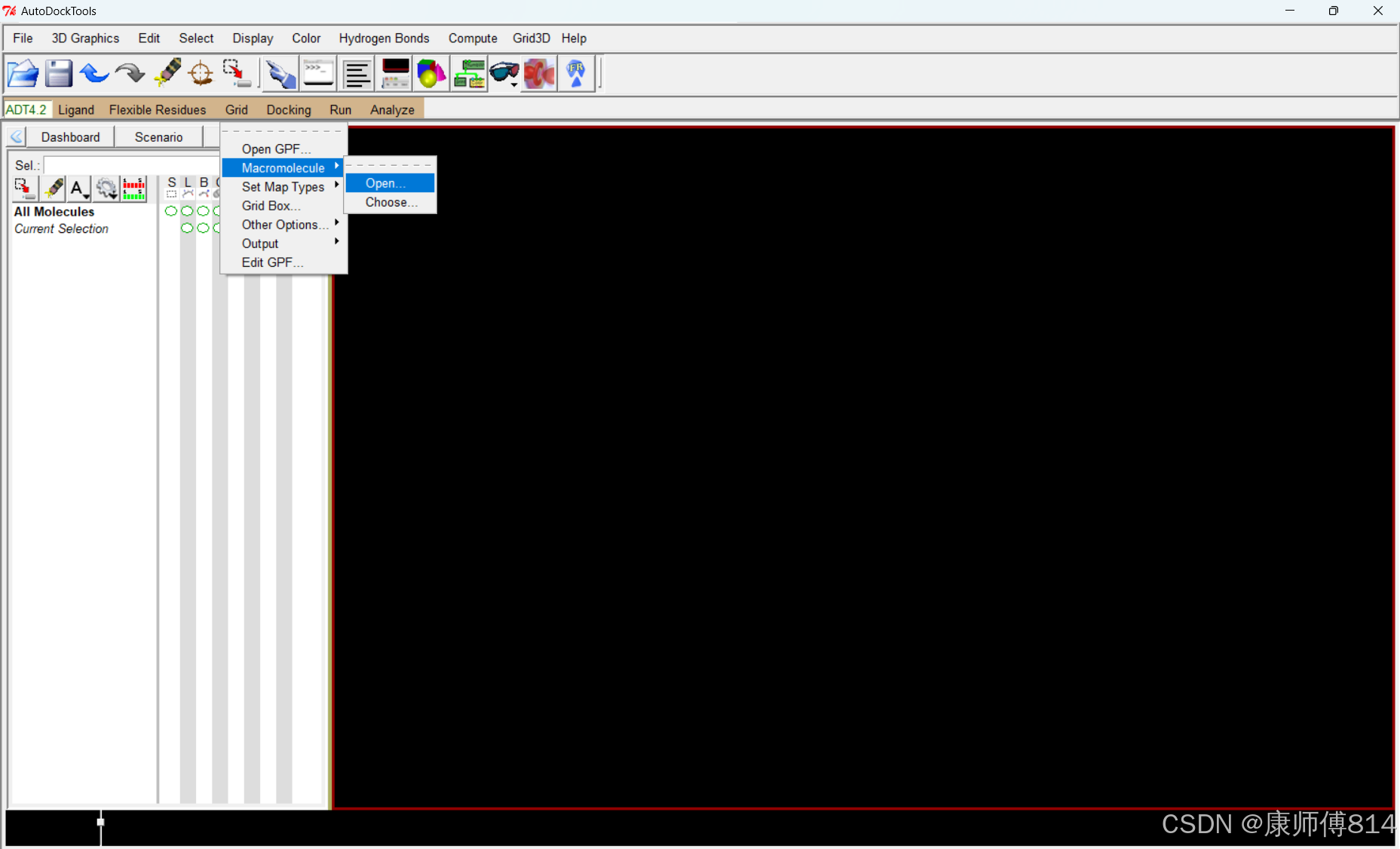
#点击Grid-Macromolecule-Open,选择蛋白质pdbqt文件
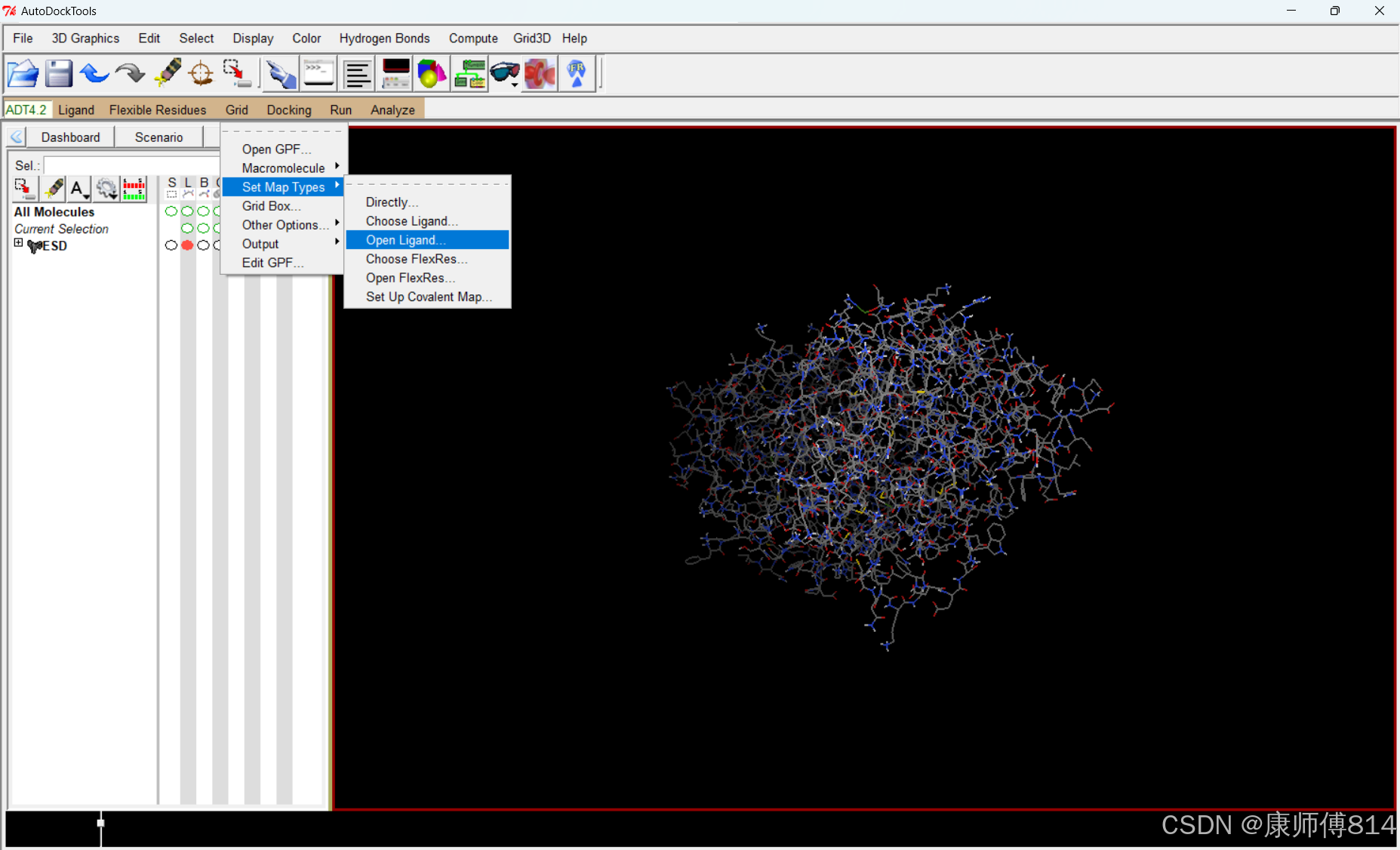
#点击Grid-Set Map Types-Open Ligand,选择小分子pdbqt文件
设置gridbox
#点击Grid-Grid box,调节gridbox大小,使得其完全覆盖蛋白质分子,但不包含小分子
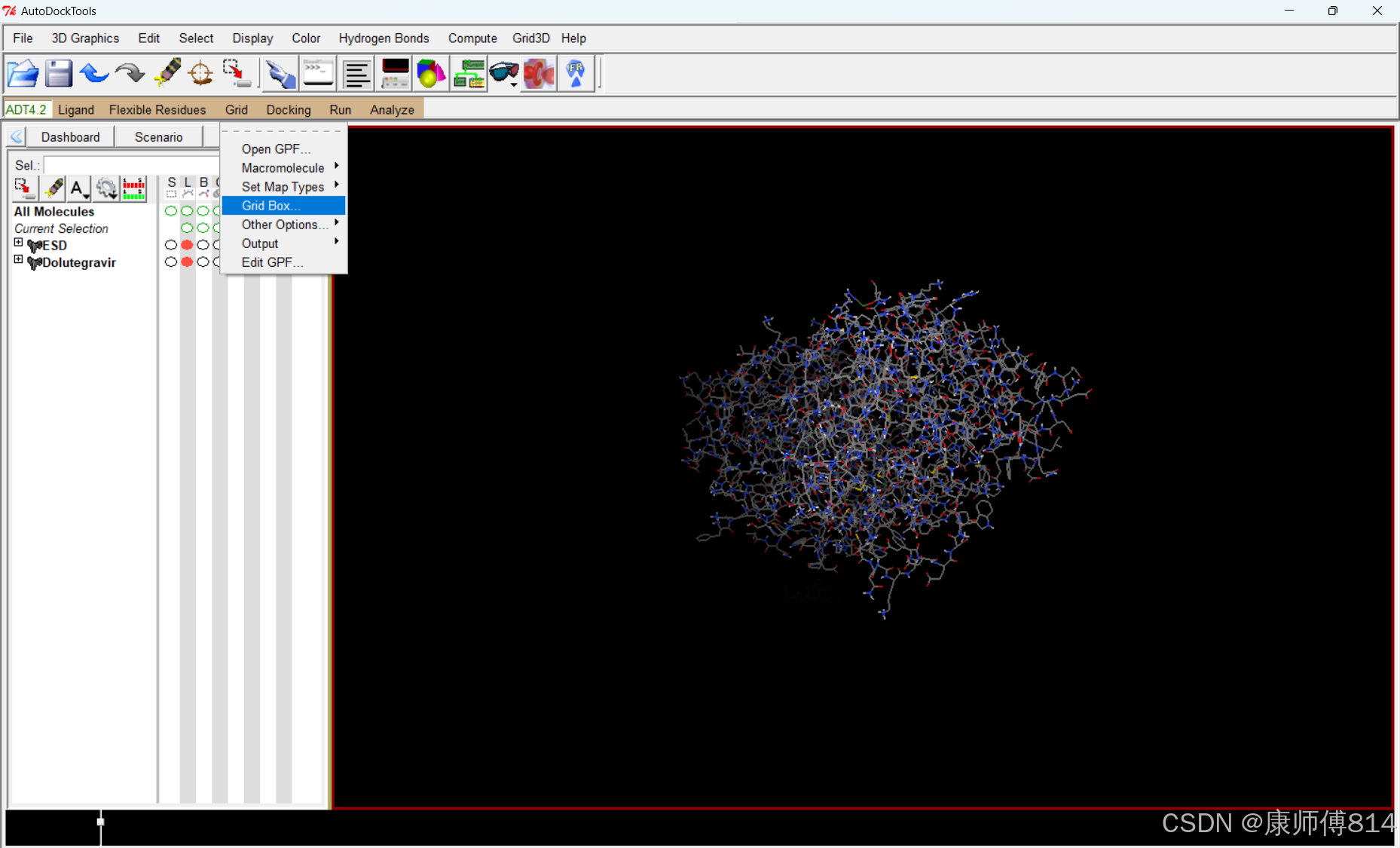
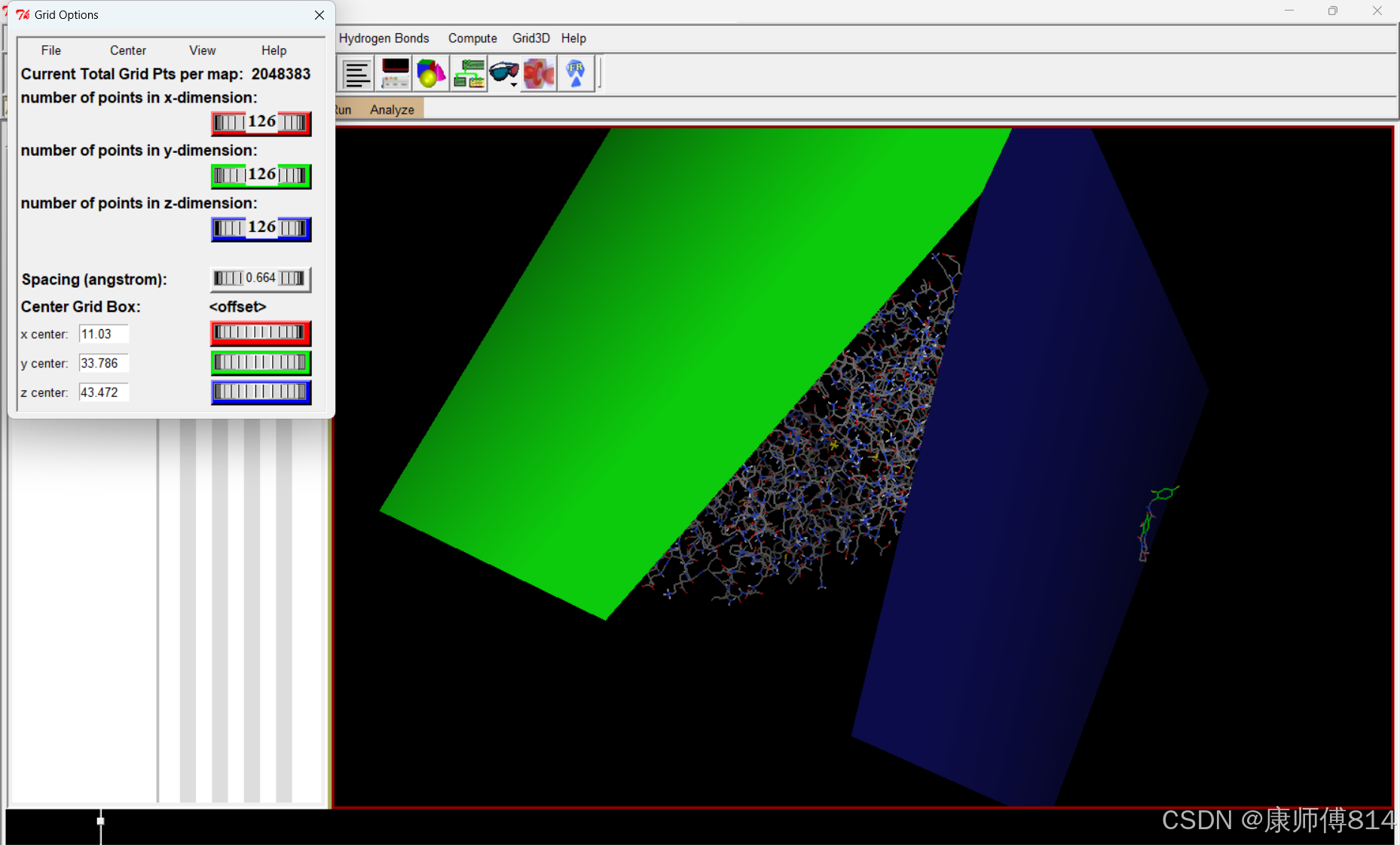
#如果确实无法做到不包含小分子结构,点击DejaVu GUI,取消勾选小分子文件的“mouse tranforms apply to "root" object only"选项,将小分子用鼠标右键拖拽出gridbox,再重新勾选该选项即可
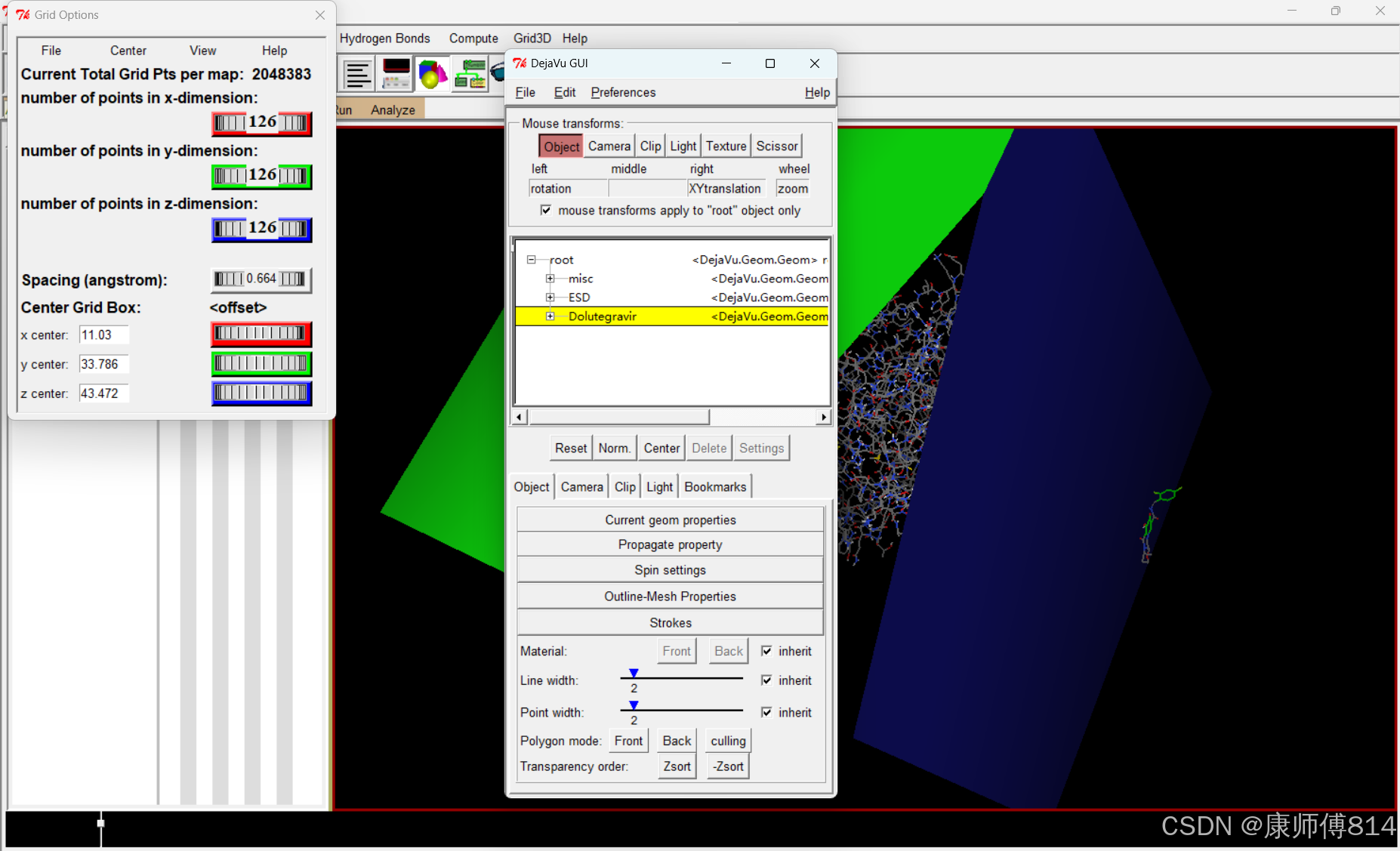
#调试好gridbox后,选择File-Close saving current即可
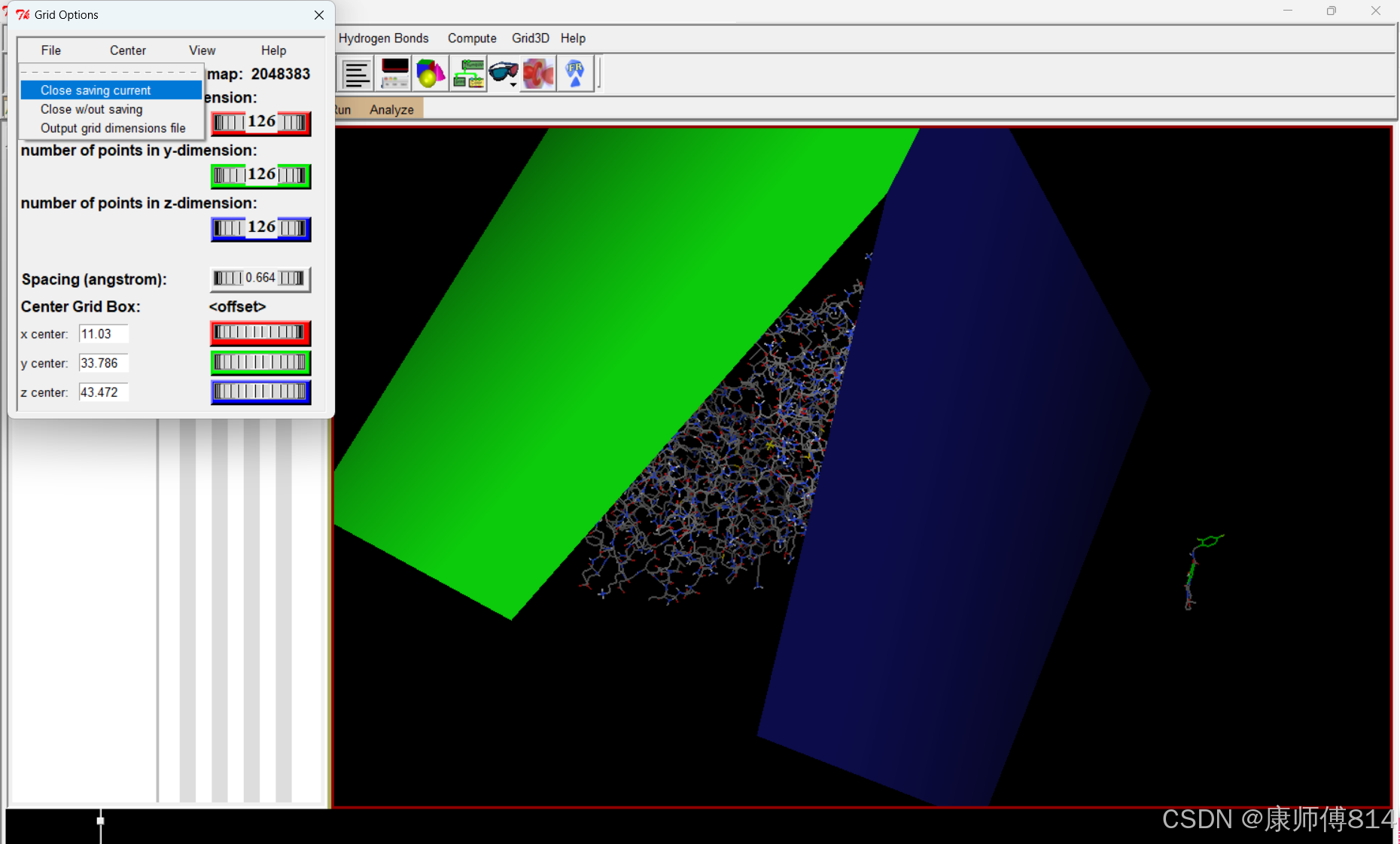
#导出为GPF
#点击Grid-Output-Save GPF,注意这里保存文件需要手动命名,带.gpf后缀,这里命名为grid.gpf
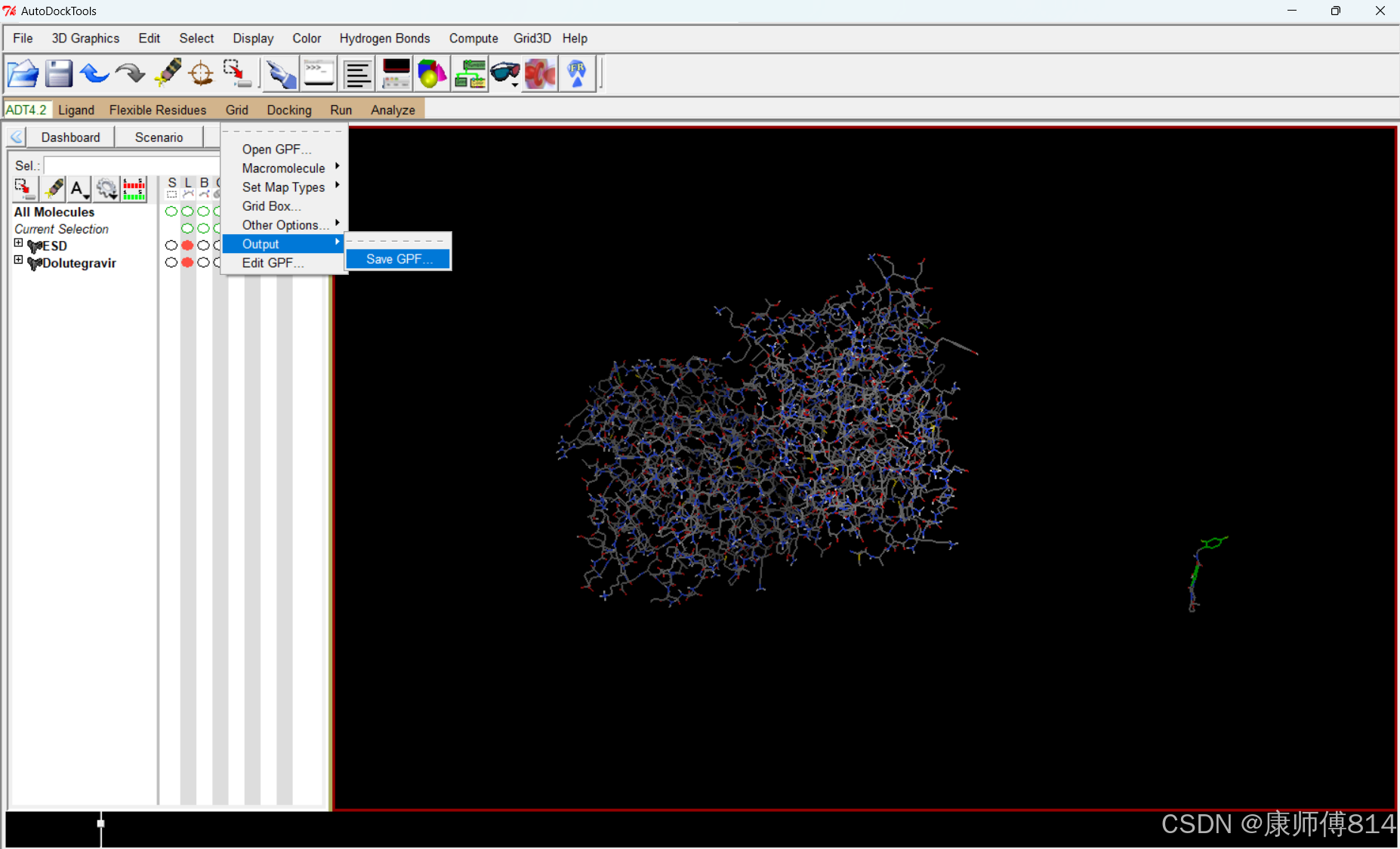
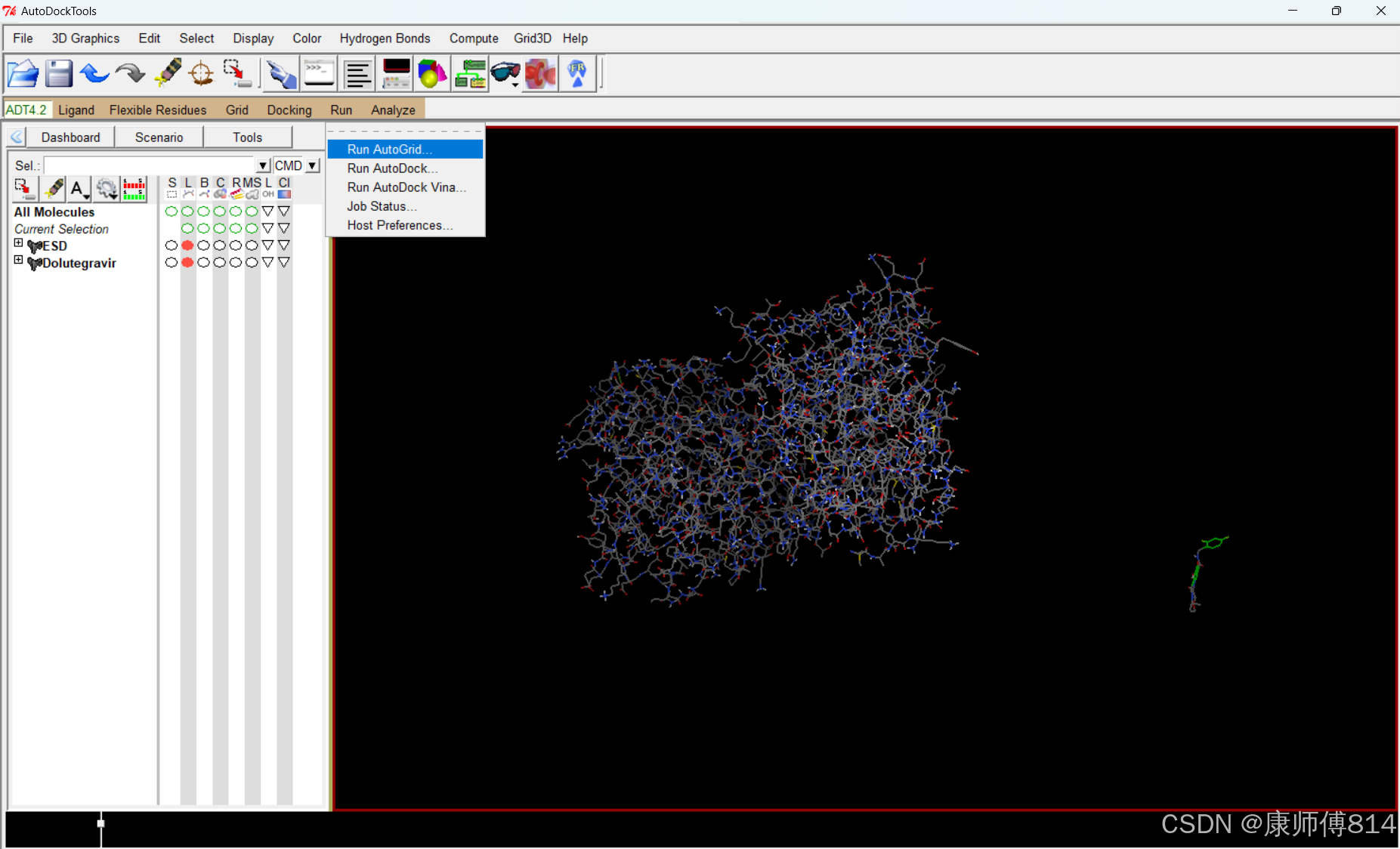
运行autogrid
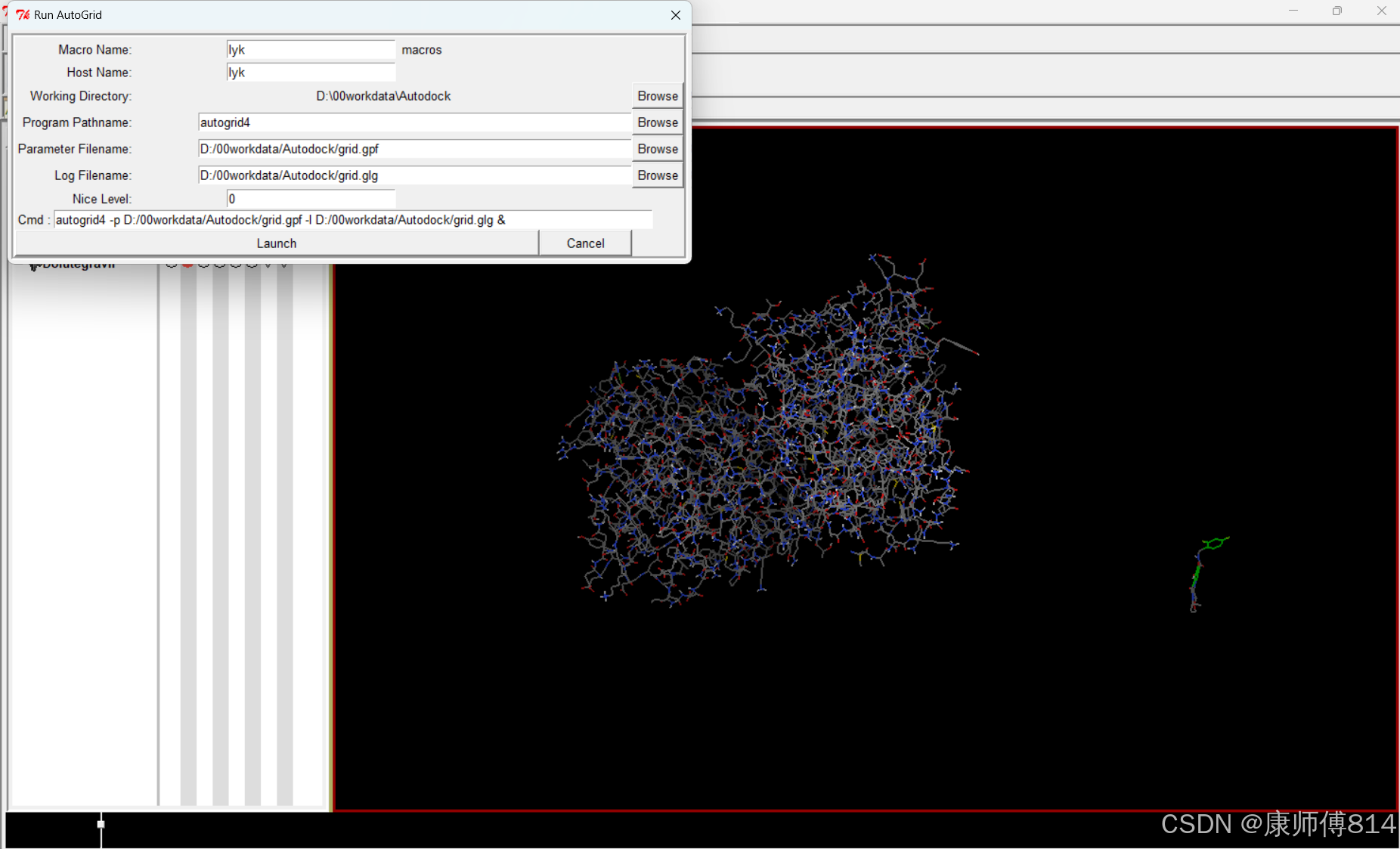
#点击Run-Run Autogrid,注意工作目录是否正确,导入刚才保存的grid.gpf文件,点击Launch,即开始运行Autogrid,约5min
#运行结束后可以看见工作目录下多了很多map文件
进行autodock
设置计算参数
#点击Docking-Macromolecule-Set Rigid Filename, 选中蛋白质的pdbqt文件
#点击Docking-Ligand-Choose,选中小分子,点击Select Ligand
#接下来的Search Parameters-Geneitc Algorithm和Docking Parameters,点击Accept选择默认参数即可
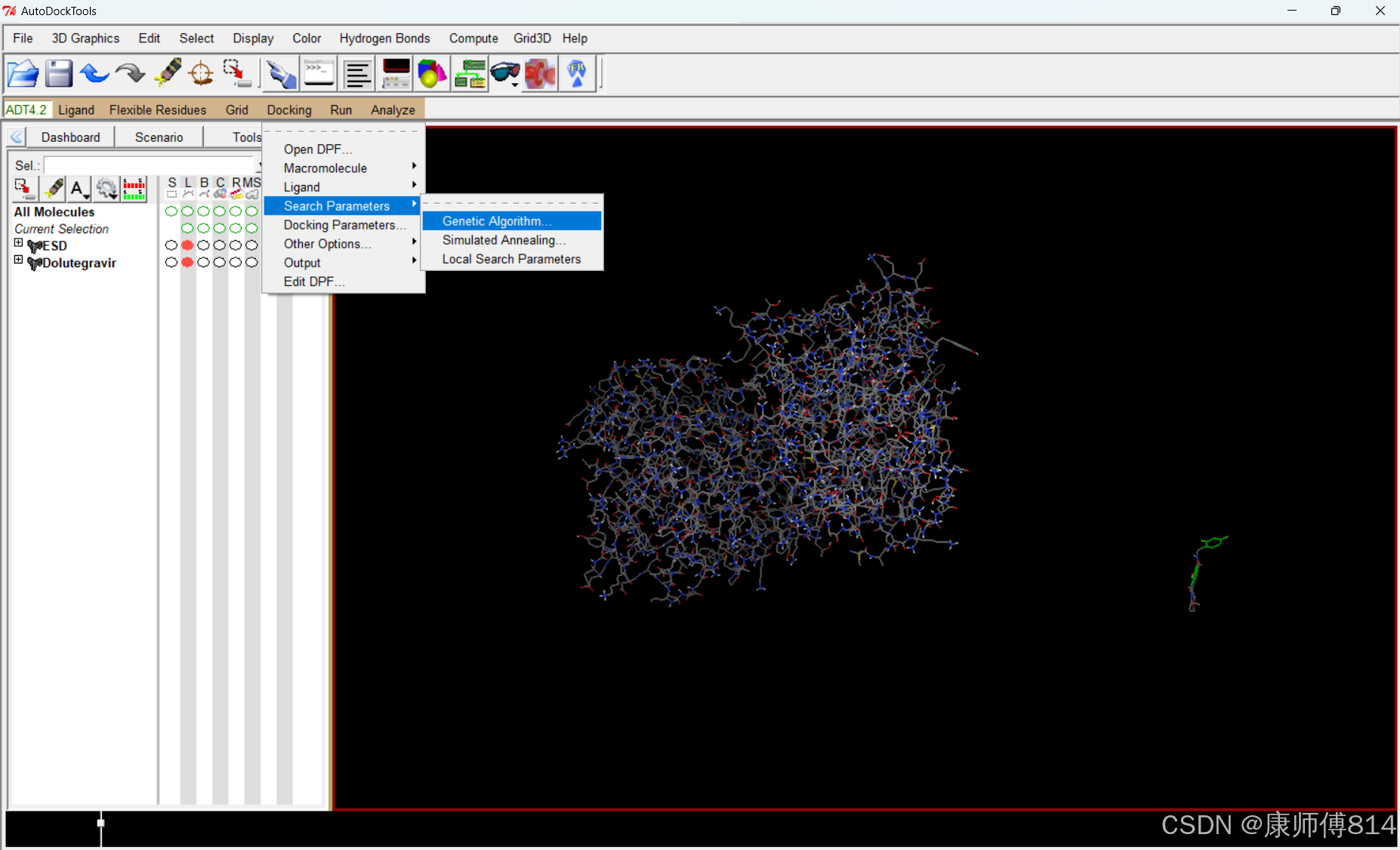
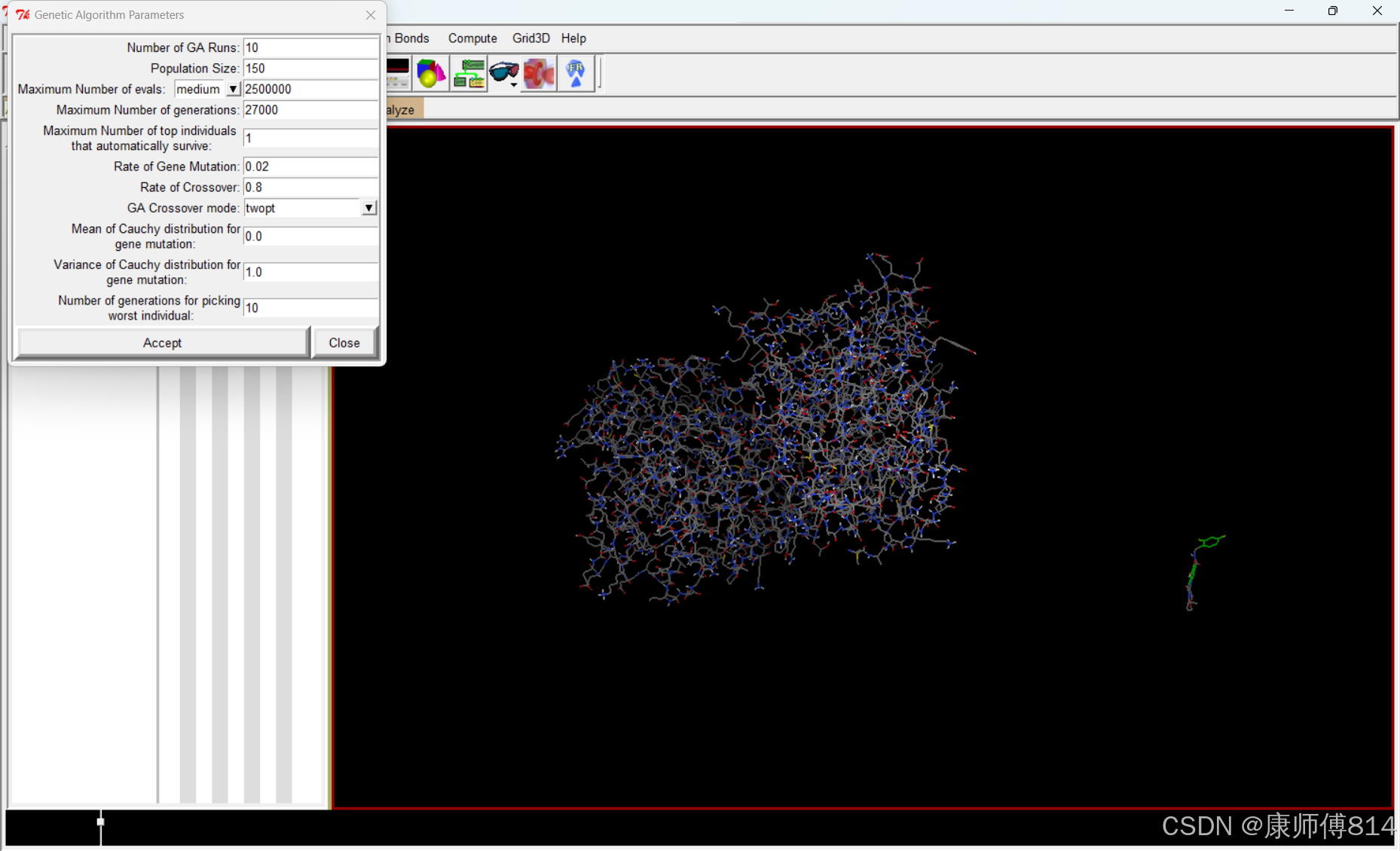
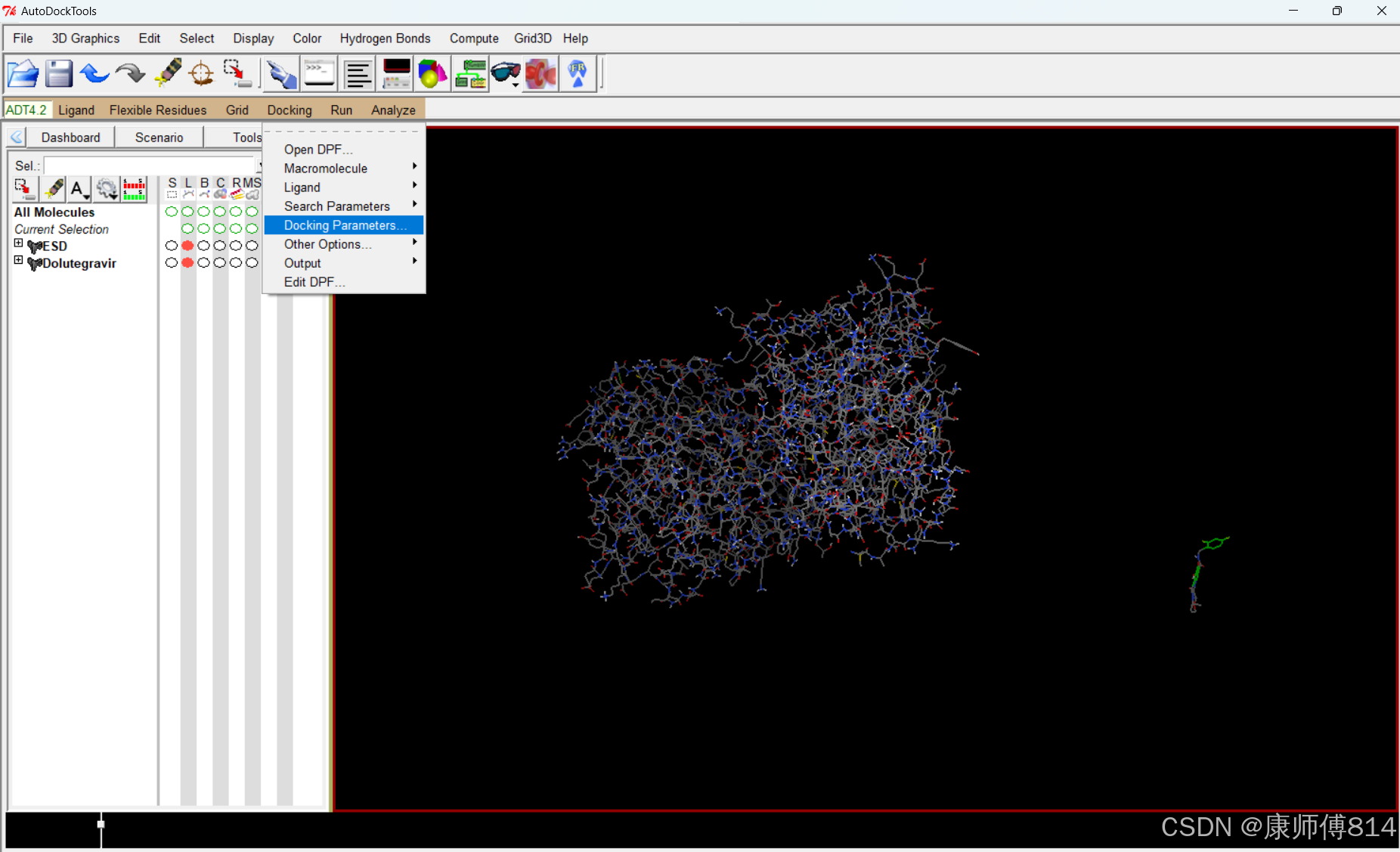
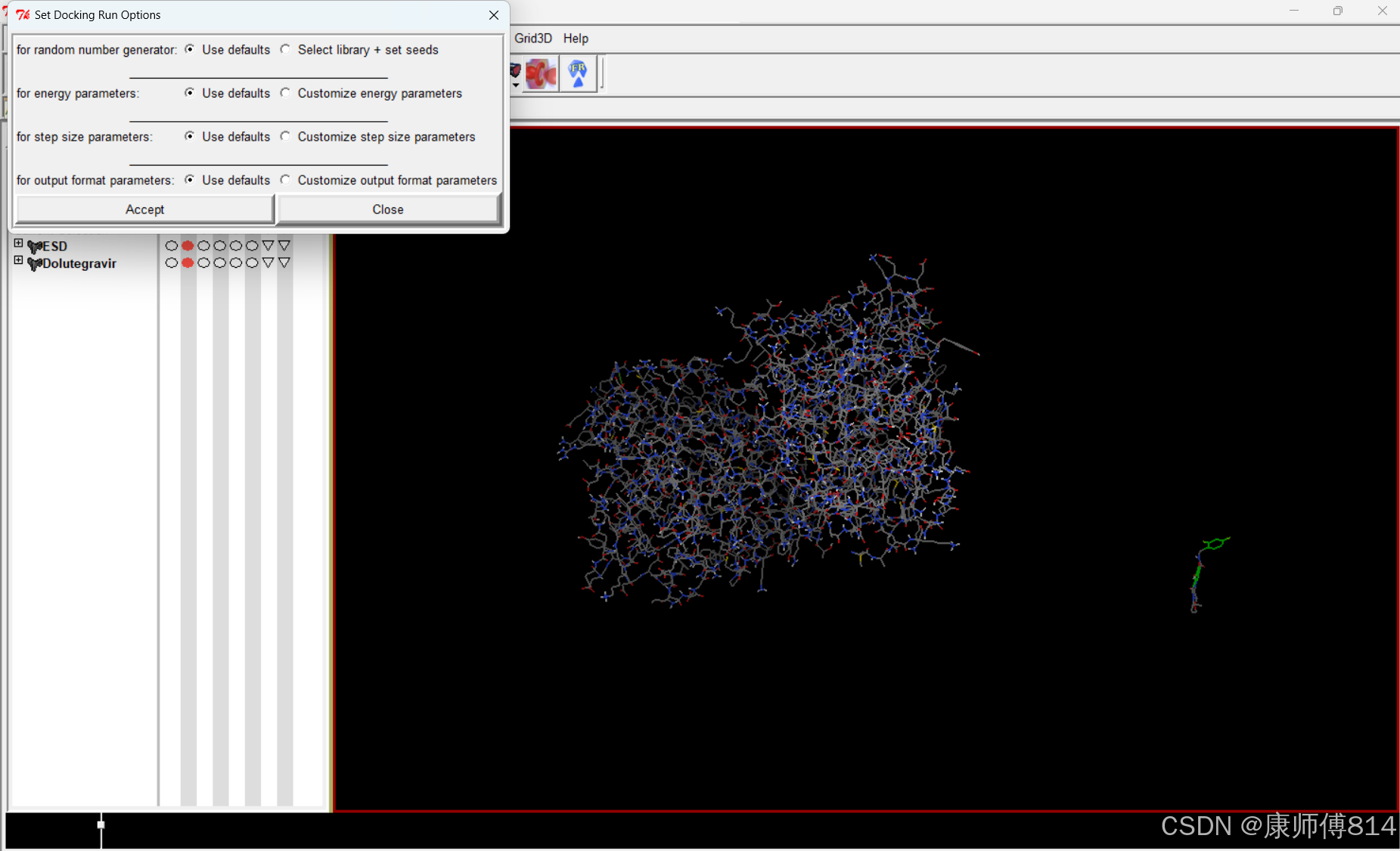
#导出设置
#点击Docking-Output-Lamarckian GA,导出为dock.dpf(此文件同样需要手动命名,如上)
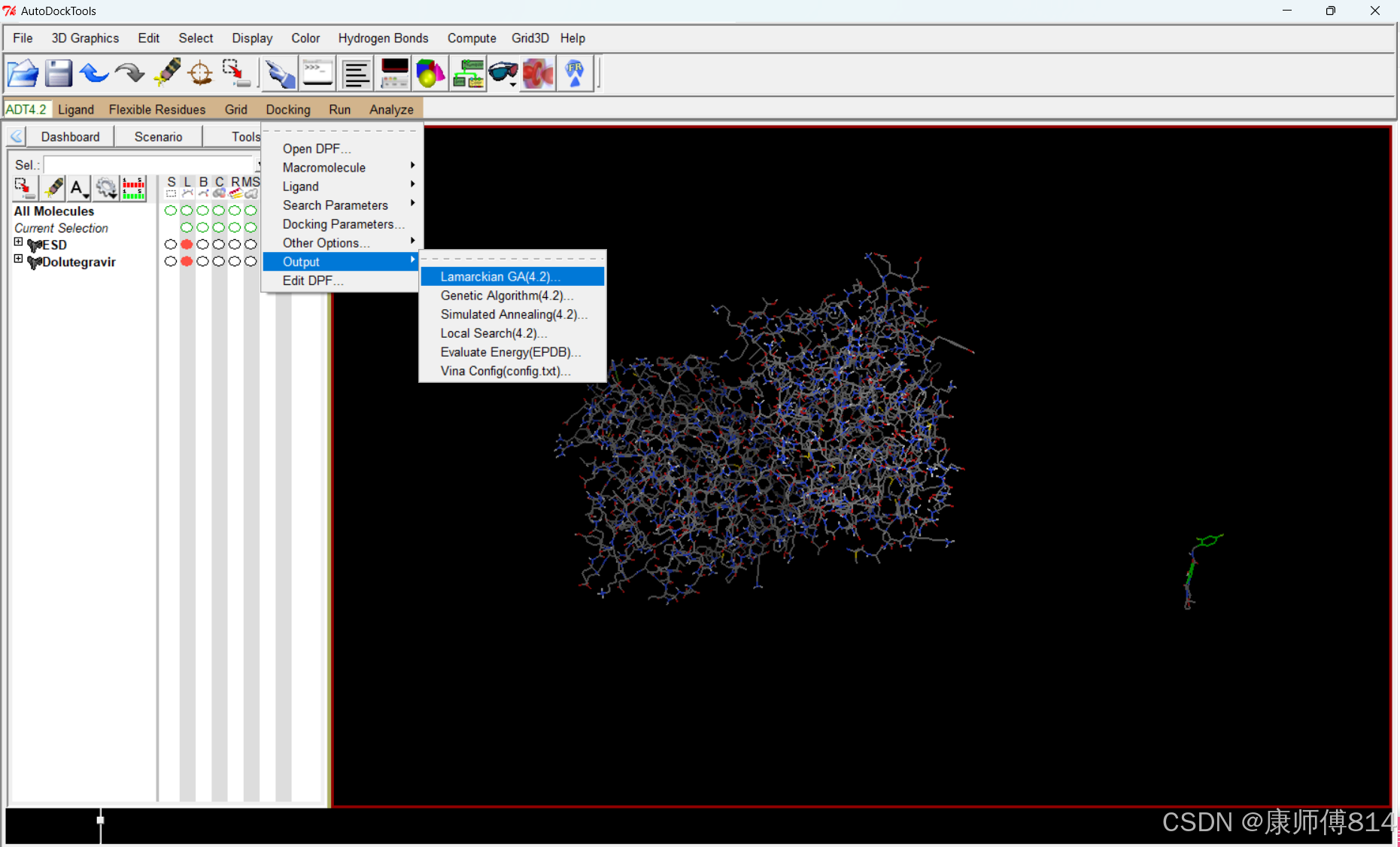
运行autodock
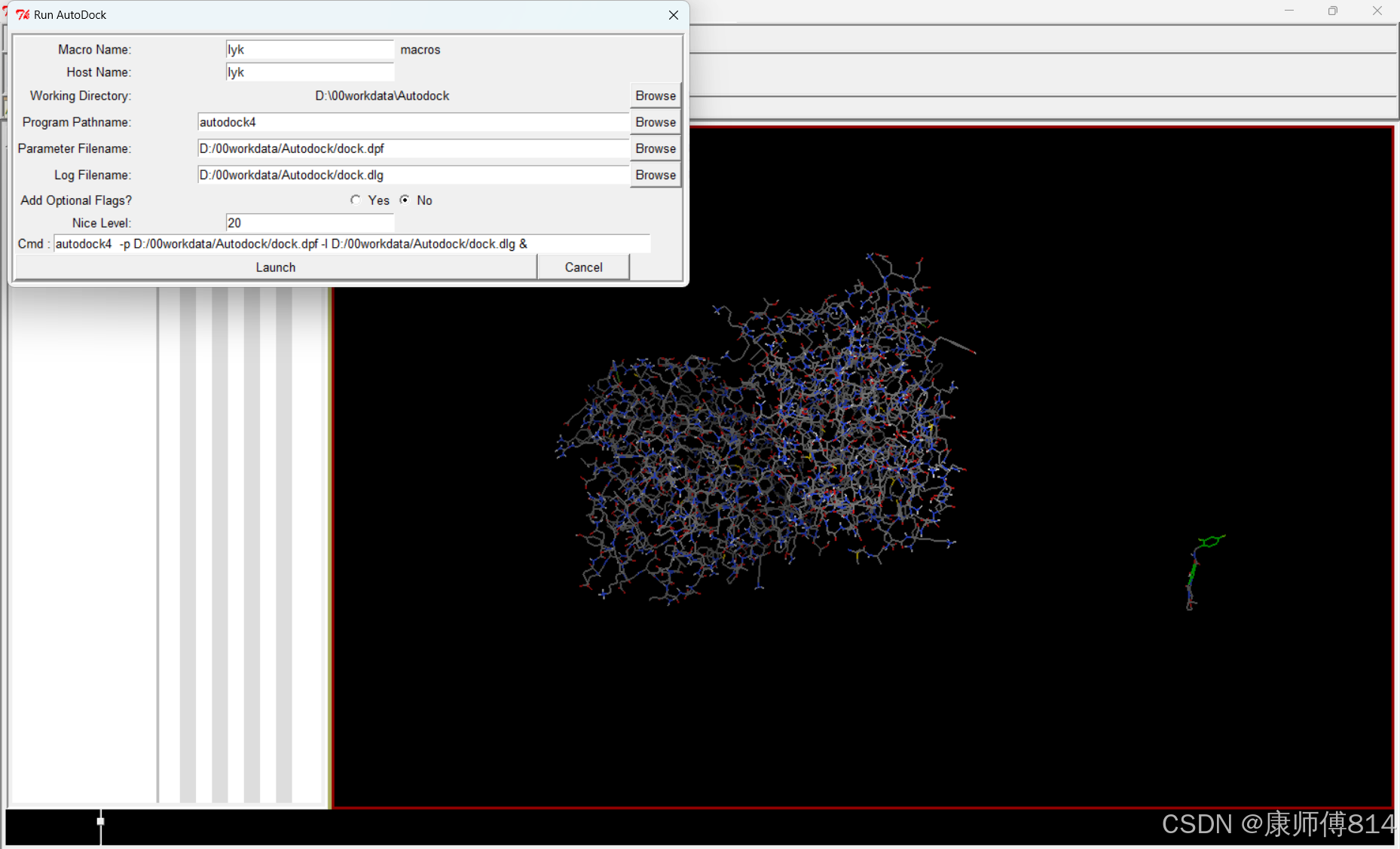
#点击Run-Run AutoDock,同样检查工作目录是否正确,上传刚才保存的dock.dpf,点击Launch,运行autodock,约10min
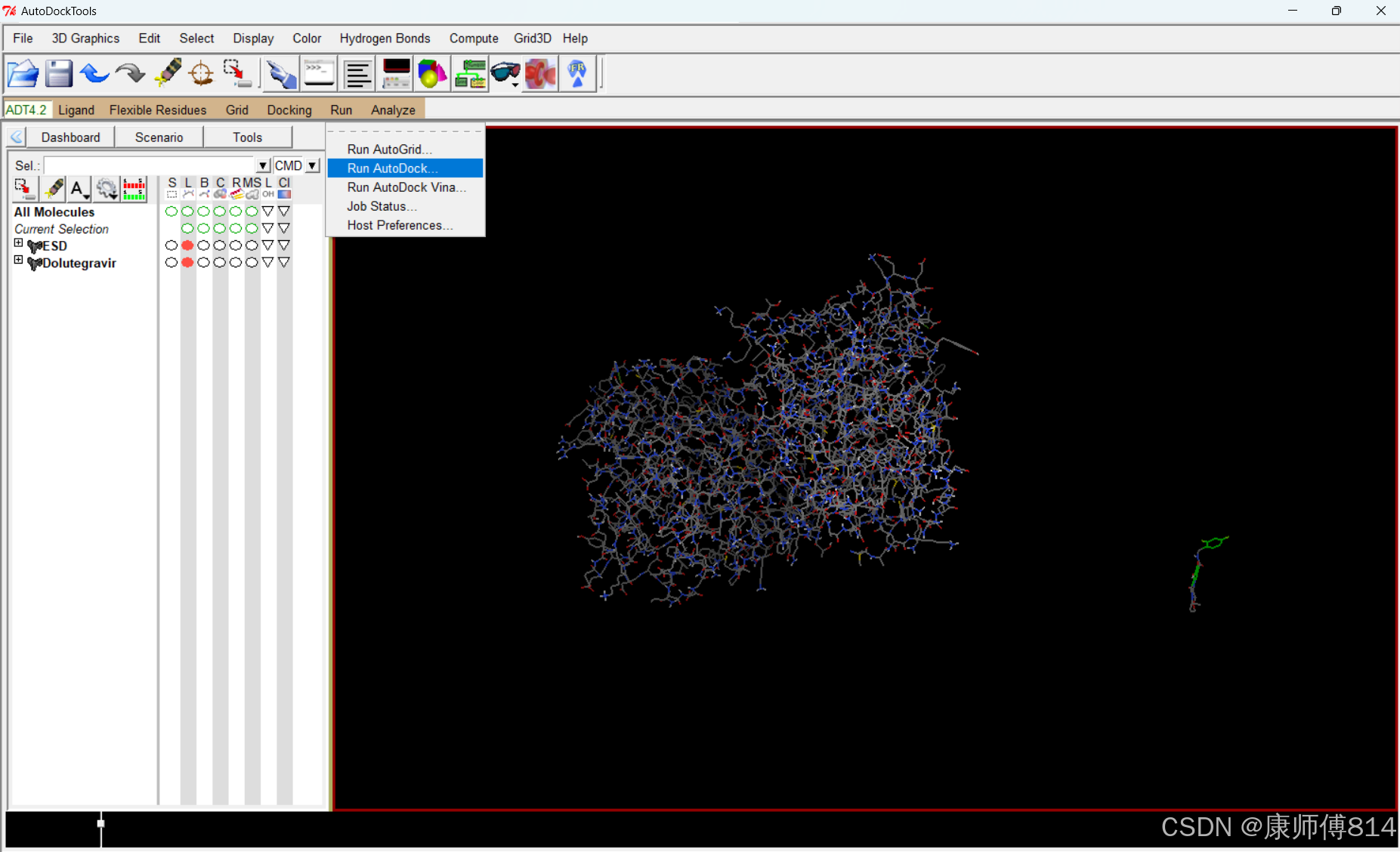
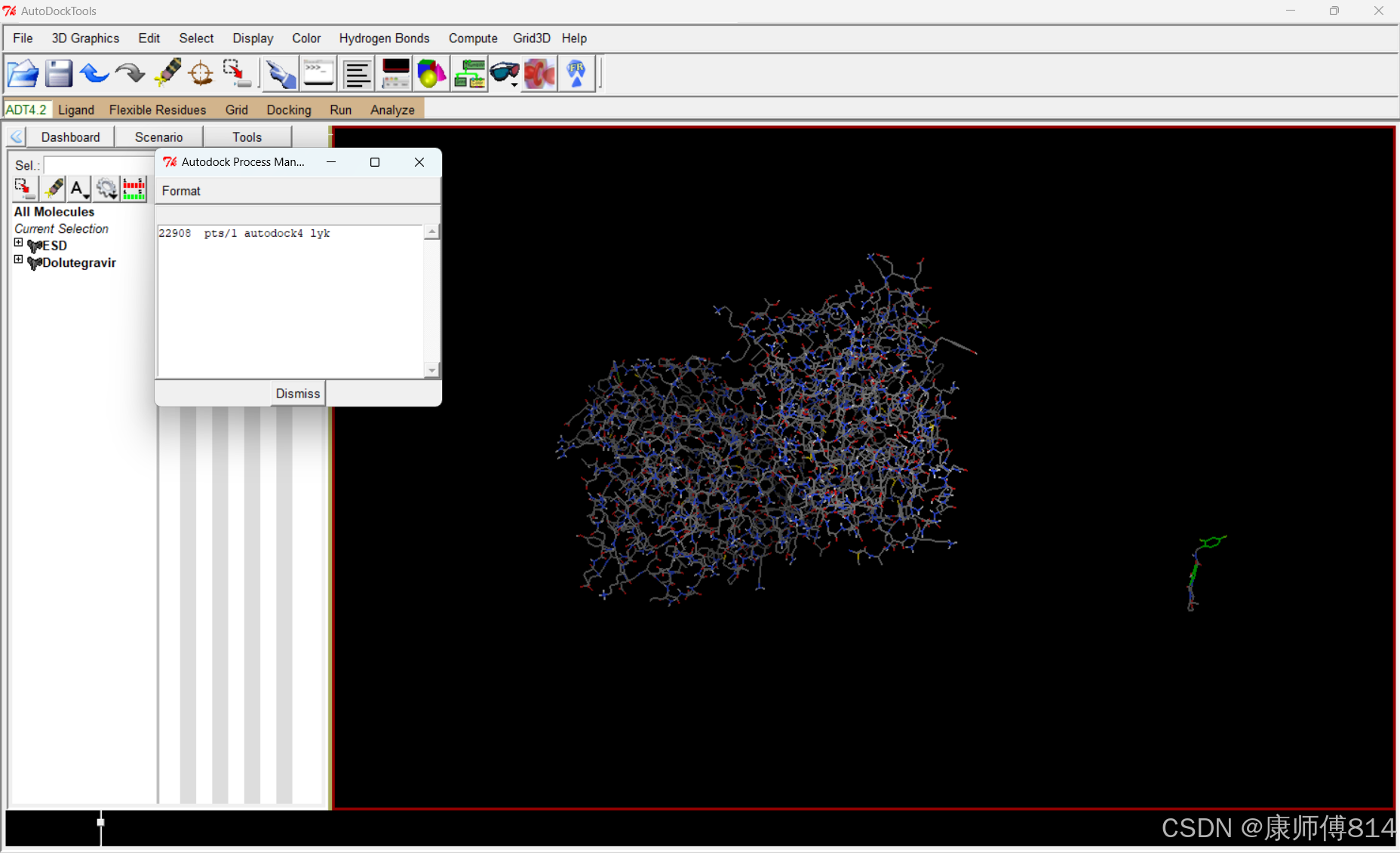
#对接完成后,查看对接结果
#删去所有分子,点击Edit-Delete-Delete All Molecules
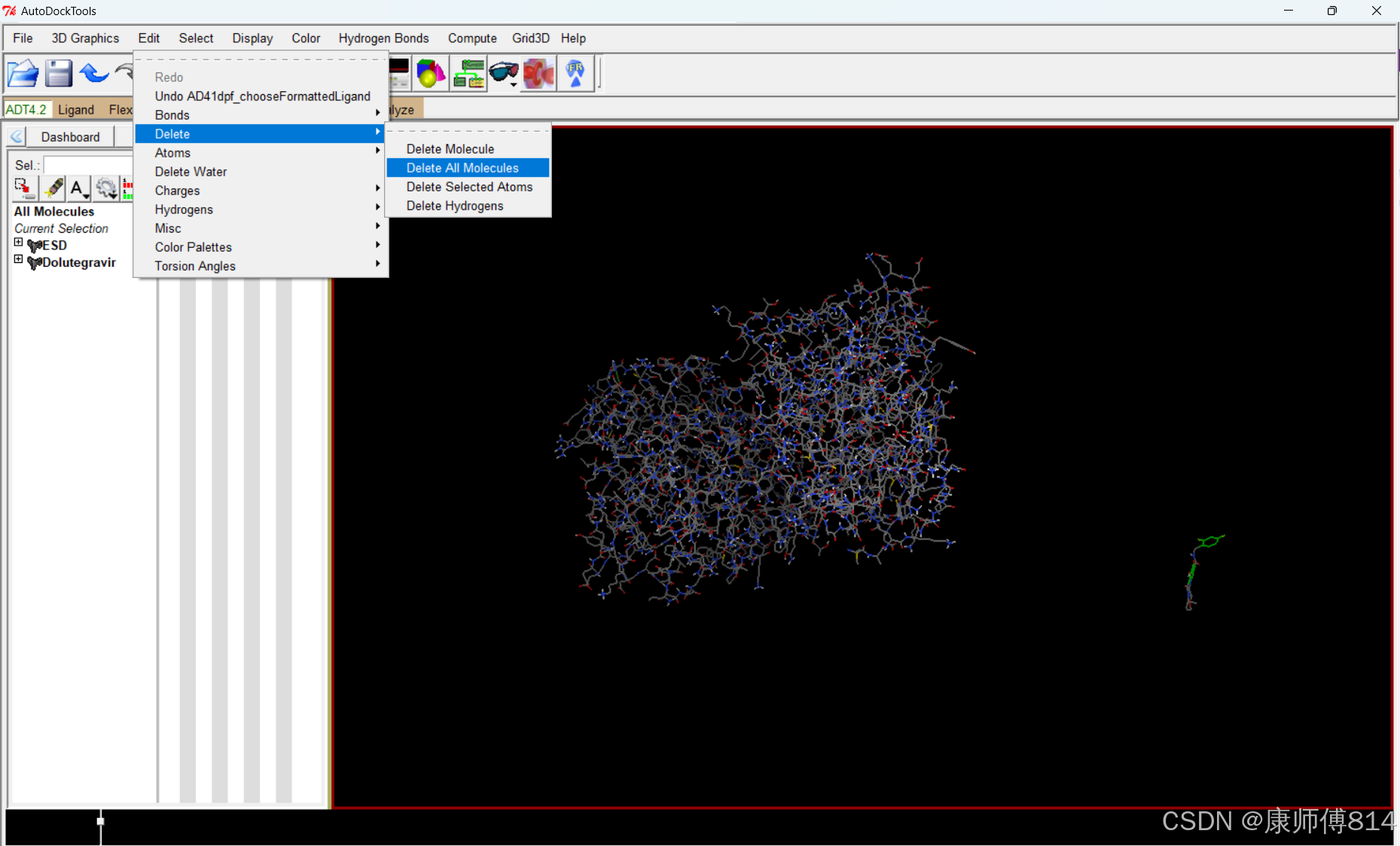
#点击Analyze-Dockings-Open,打开后缀为.dlg的结果文件
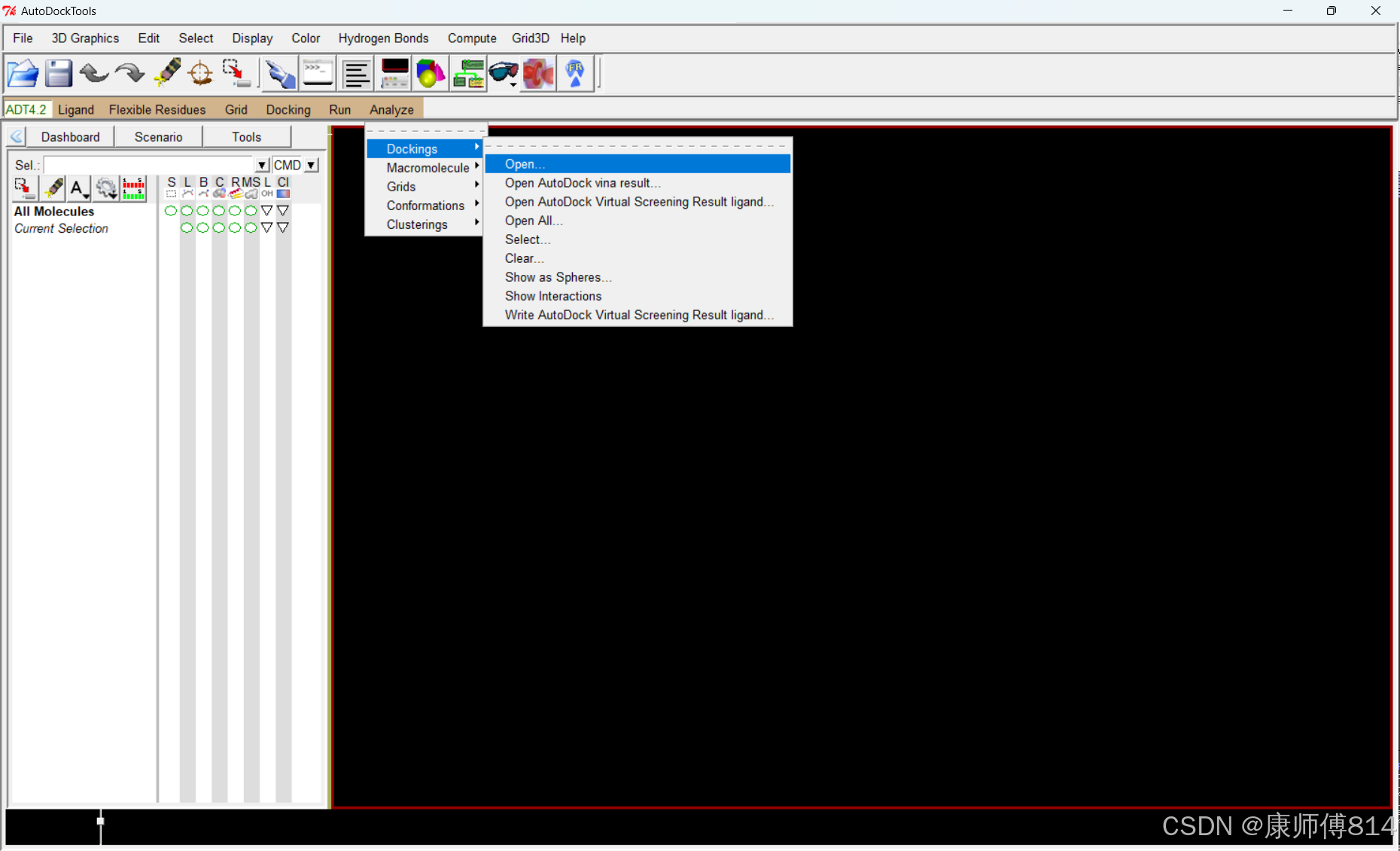
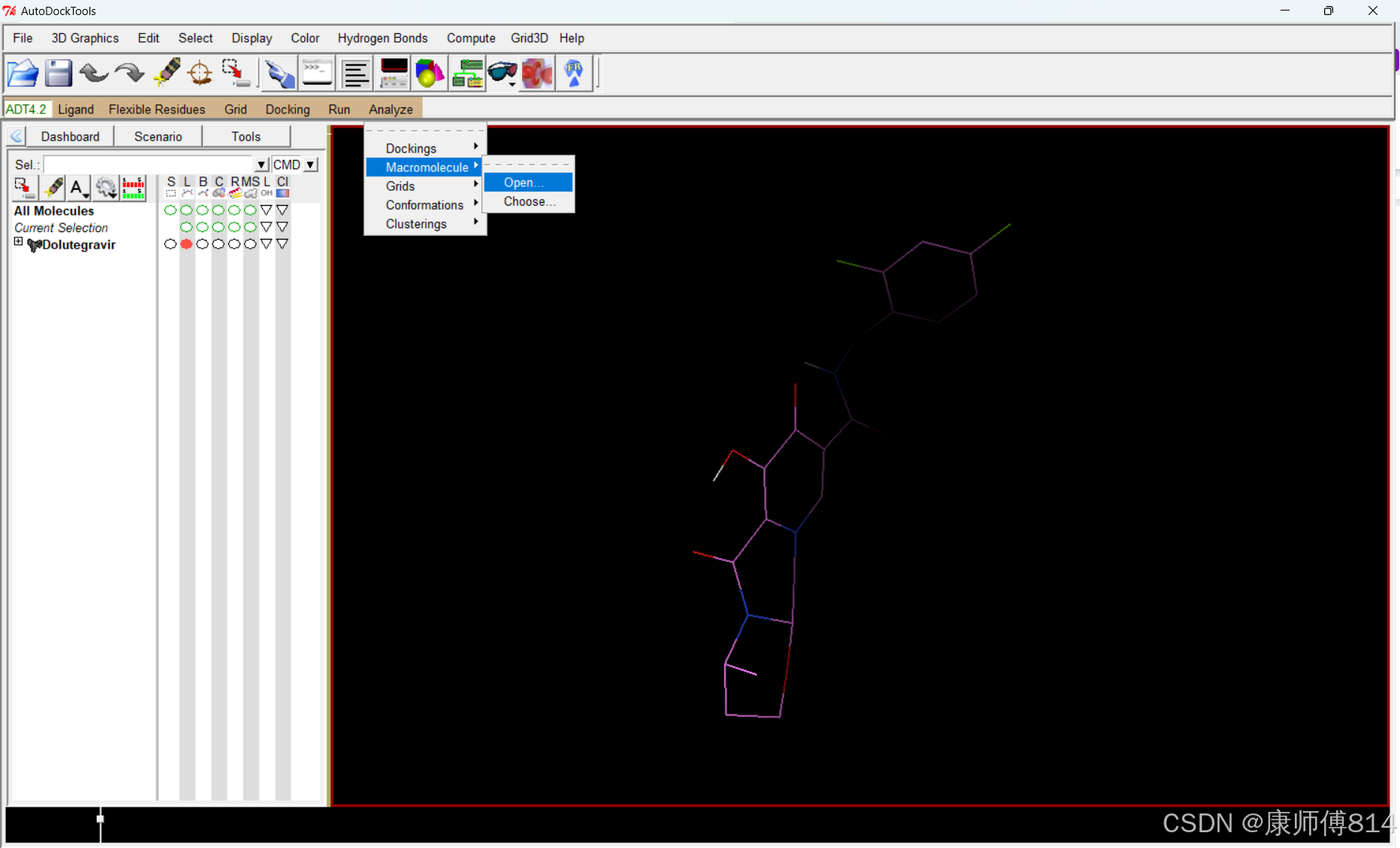
#点击Analyze-Macromolecule-Open打开分子结构
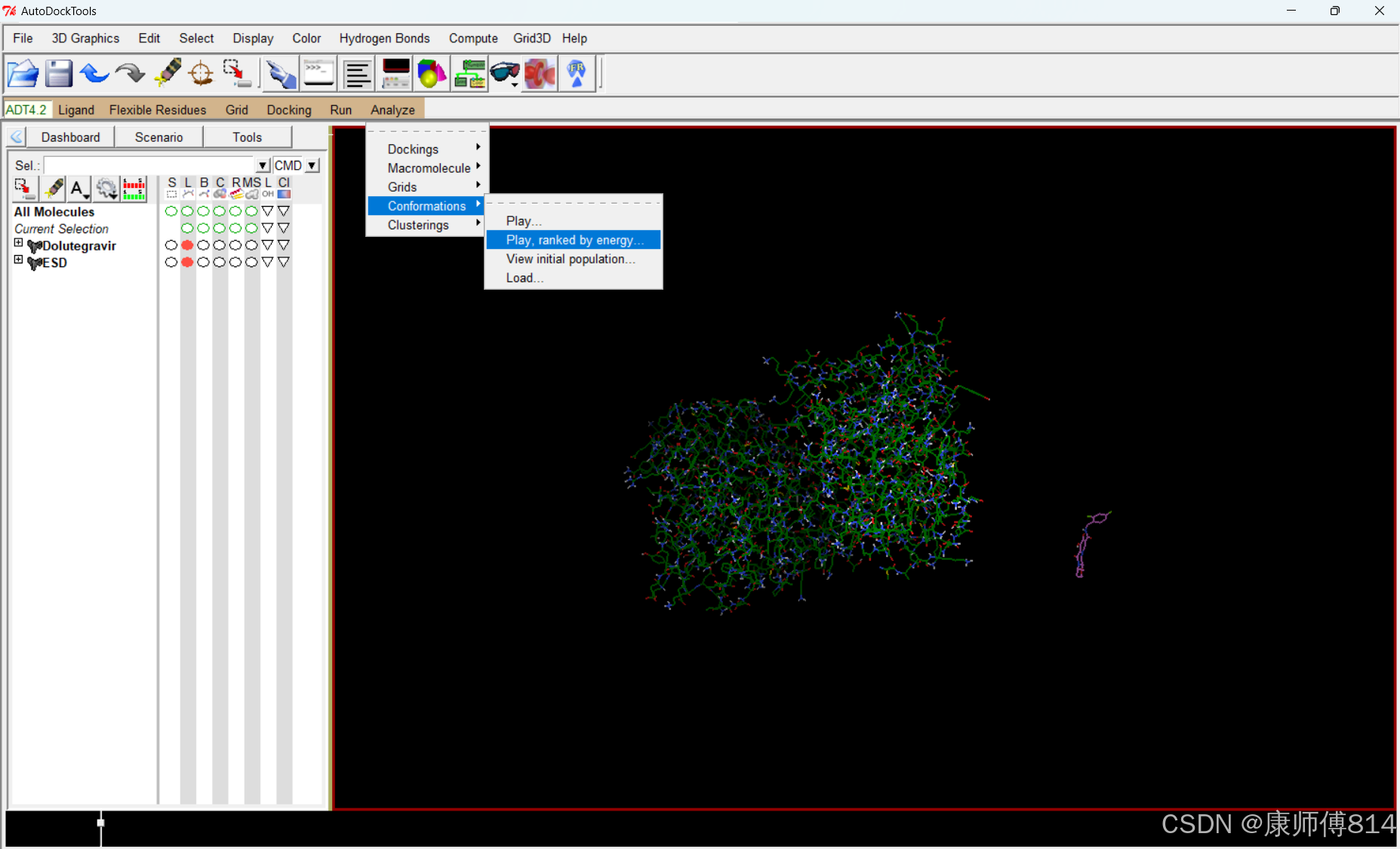
#点击Analyze-Conformations-Play ranked bu enegy,可按照结合自由能从最小到最大顺序观看结合状态(默认模拟对接10次)
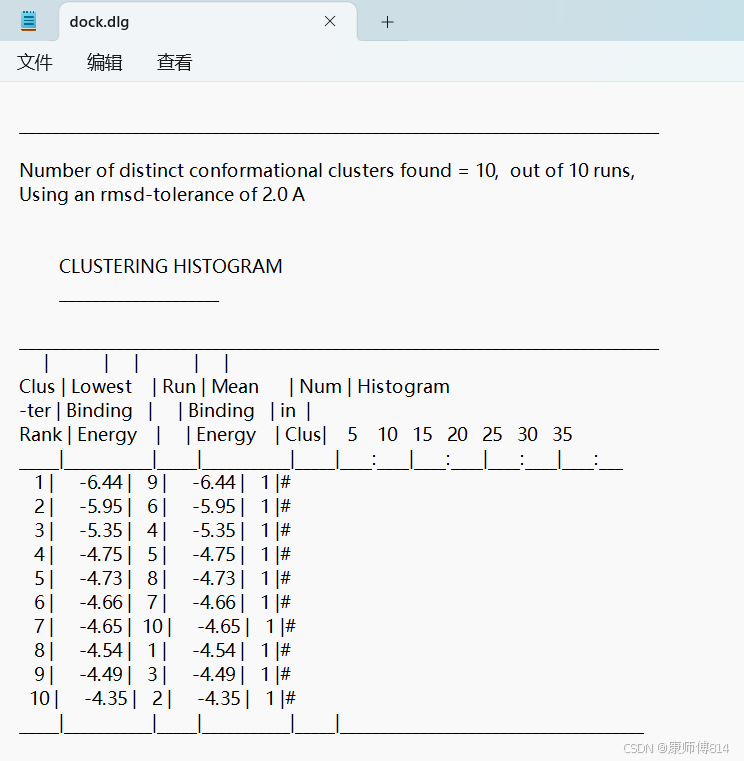
#另外,直接用记事本打开.dlg文件也可以直接看到所有对接结果的结合自由能
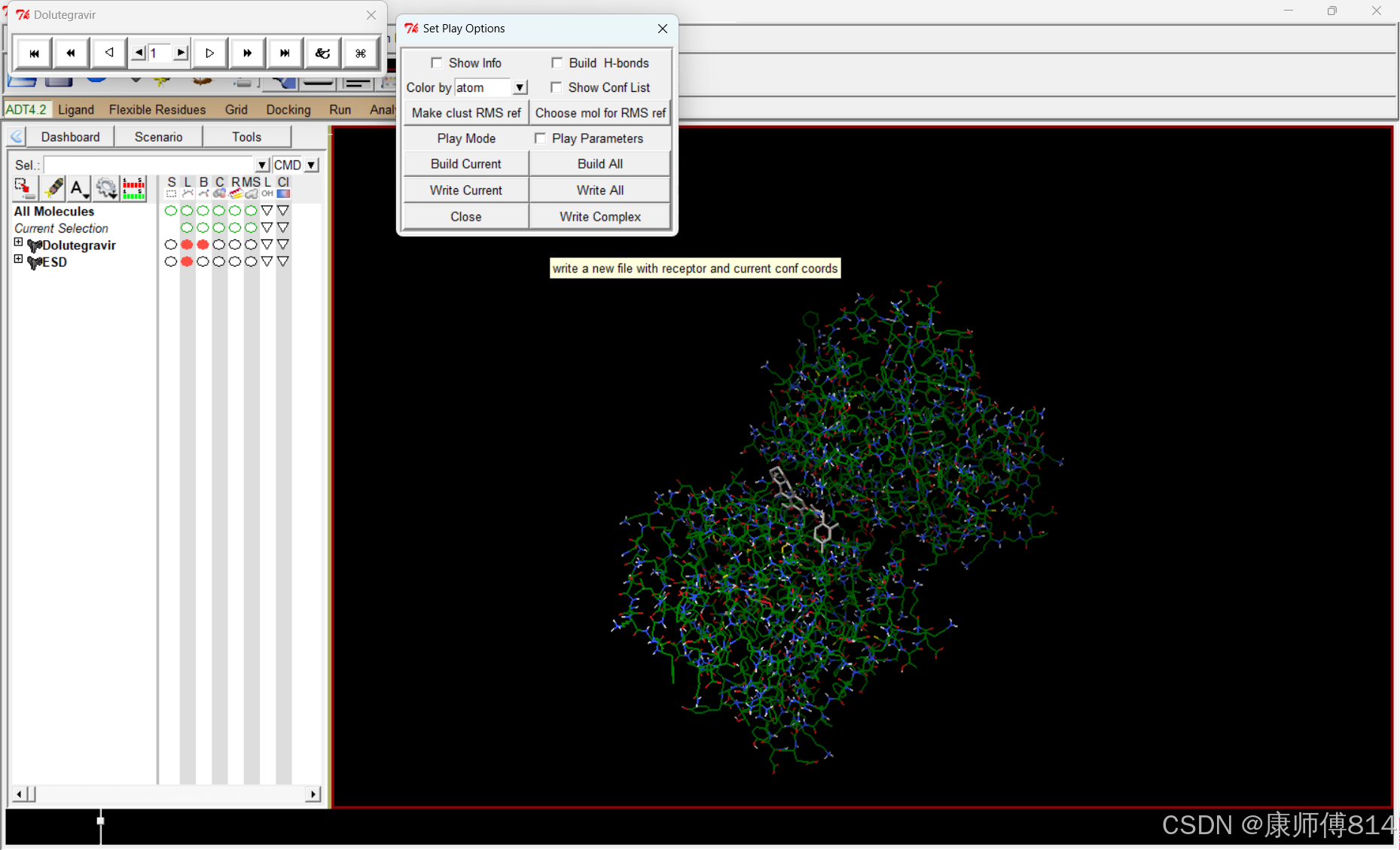
导出对接结果
#点击Set Play Options-Whie Complex导出选中的对接状态为pdbqt文件,这里我导出为ESD-Dolutegravir.pdbqt
可视化
#pymol不能直接打开pdbqt文件,所以要先将其转化为pdb文件
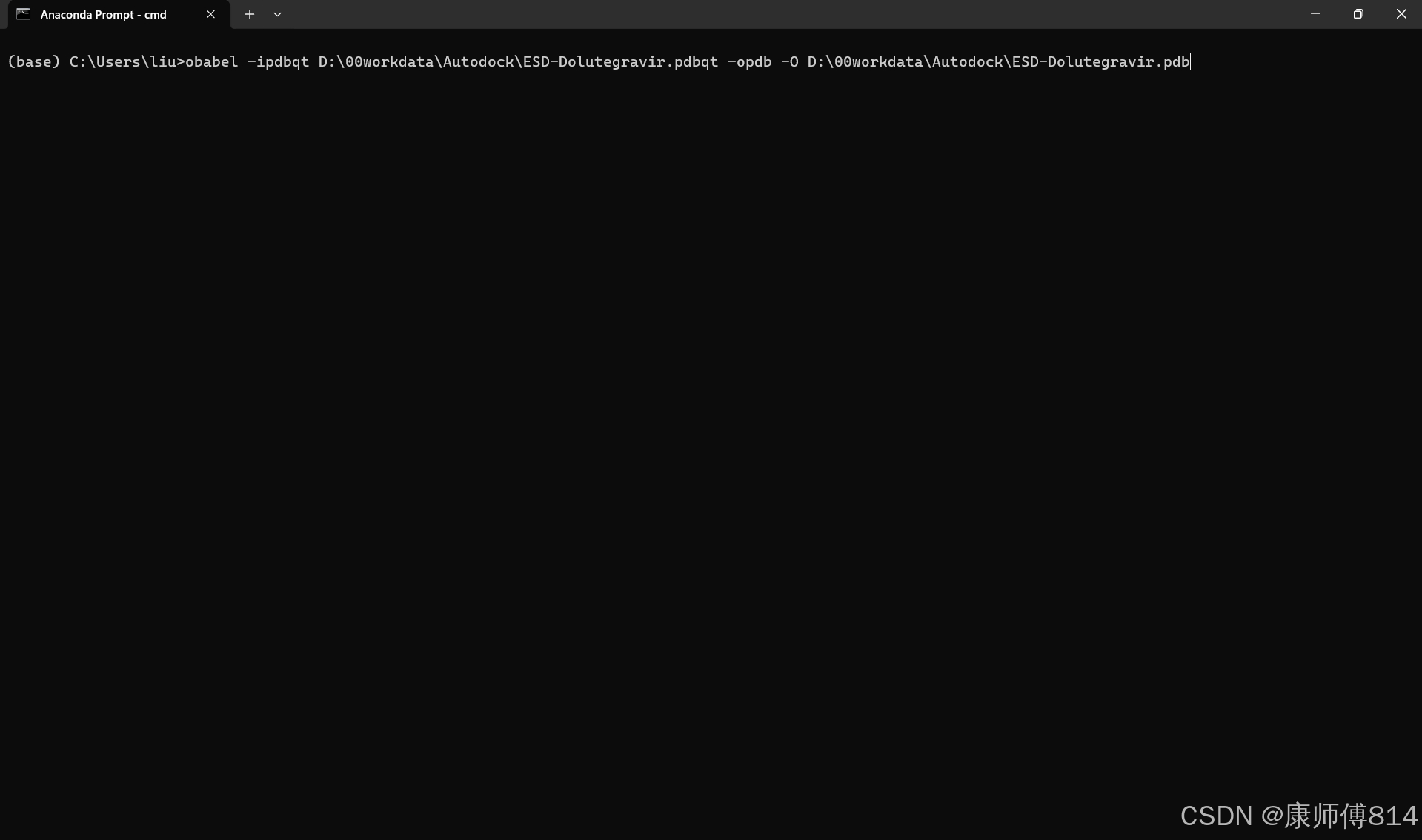
#在Anaconda Prompt打开openbabel,输入obabel -H打开帮助页面
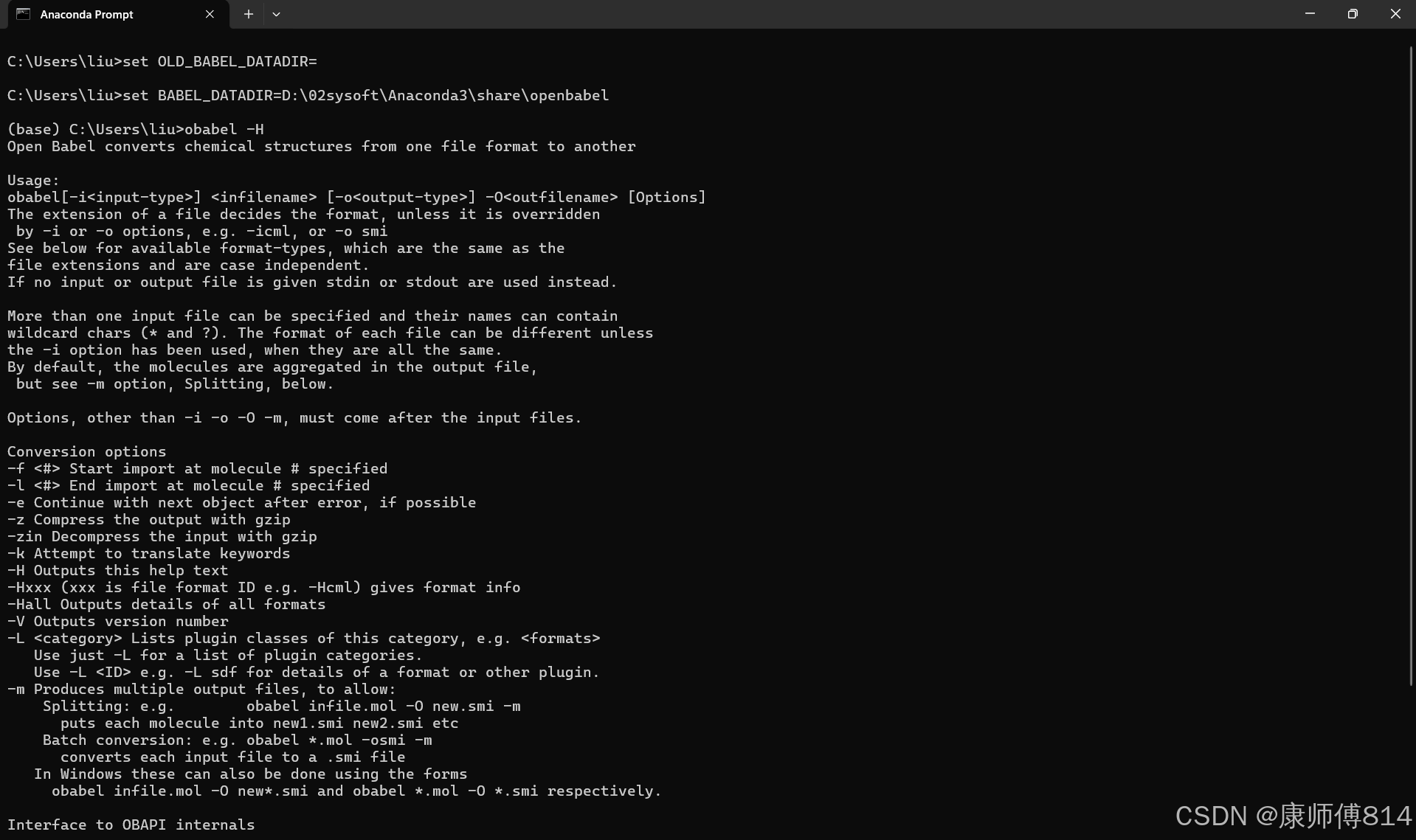
#可以看到,openbabel命令格式为obabel[-i<input-type>] <infilename> [-o<output-type>] -O<outfilename> [Options]
#将命令中的文件和路径替换为我们的文件和路径即可
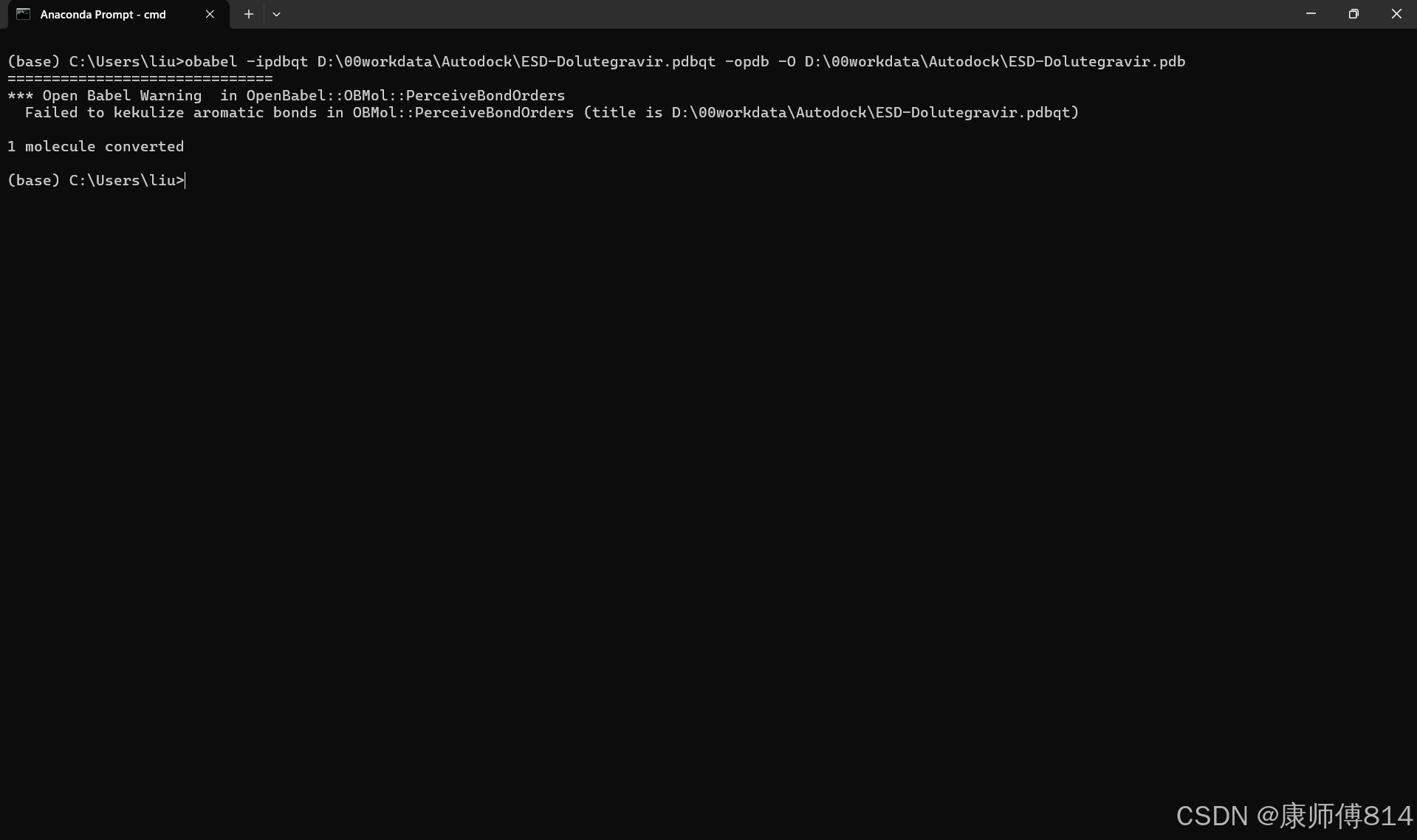
#因此,输入obabel -ipdbqt D:\00workdata\Autodock\ESD-Dolutegravir.pdbqt -opdb -O D:\00workdata\Autodock\ESD-Dolutegravir.pdb
#接下来打开pymol操作
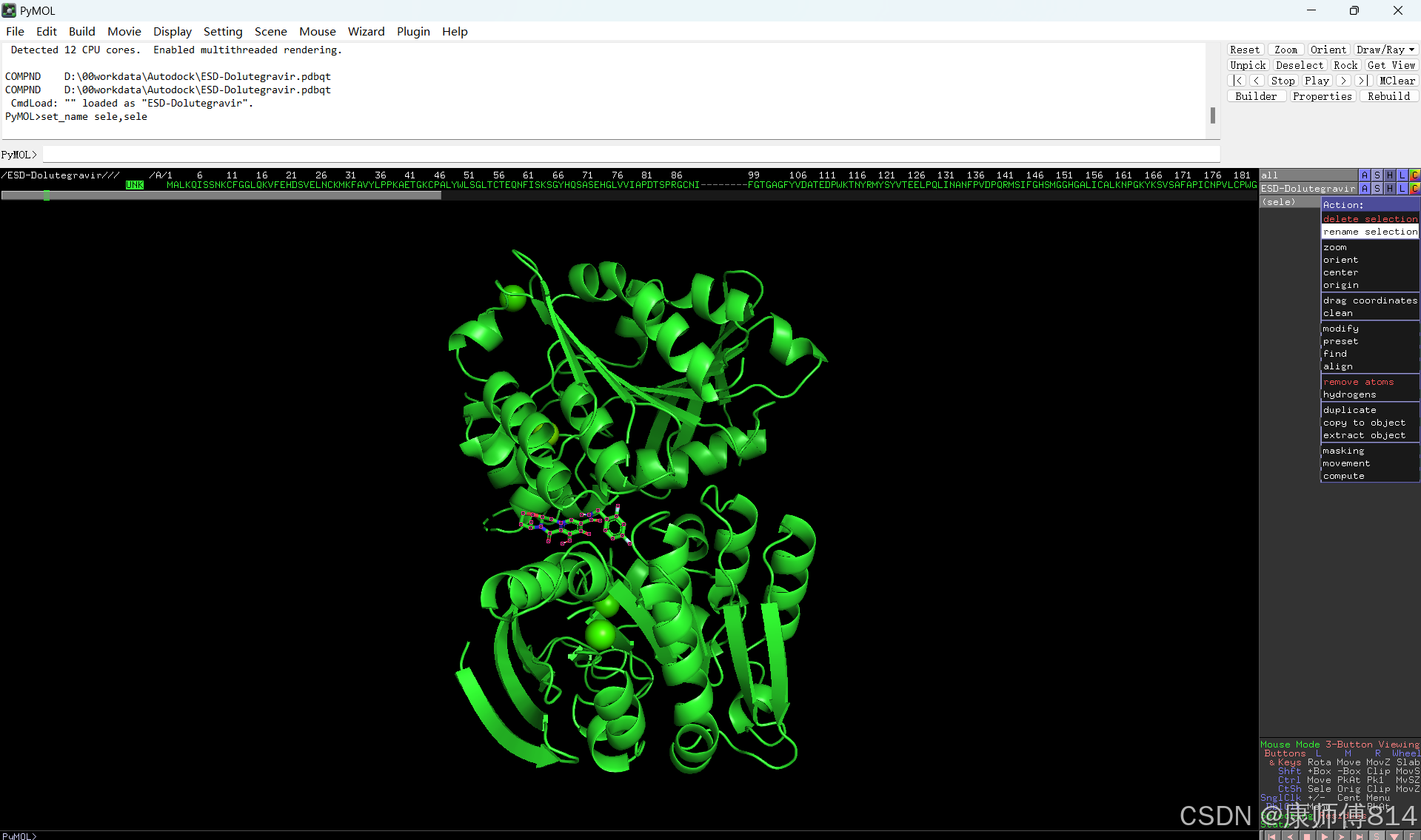
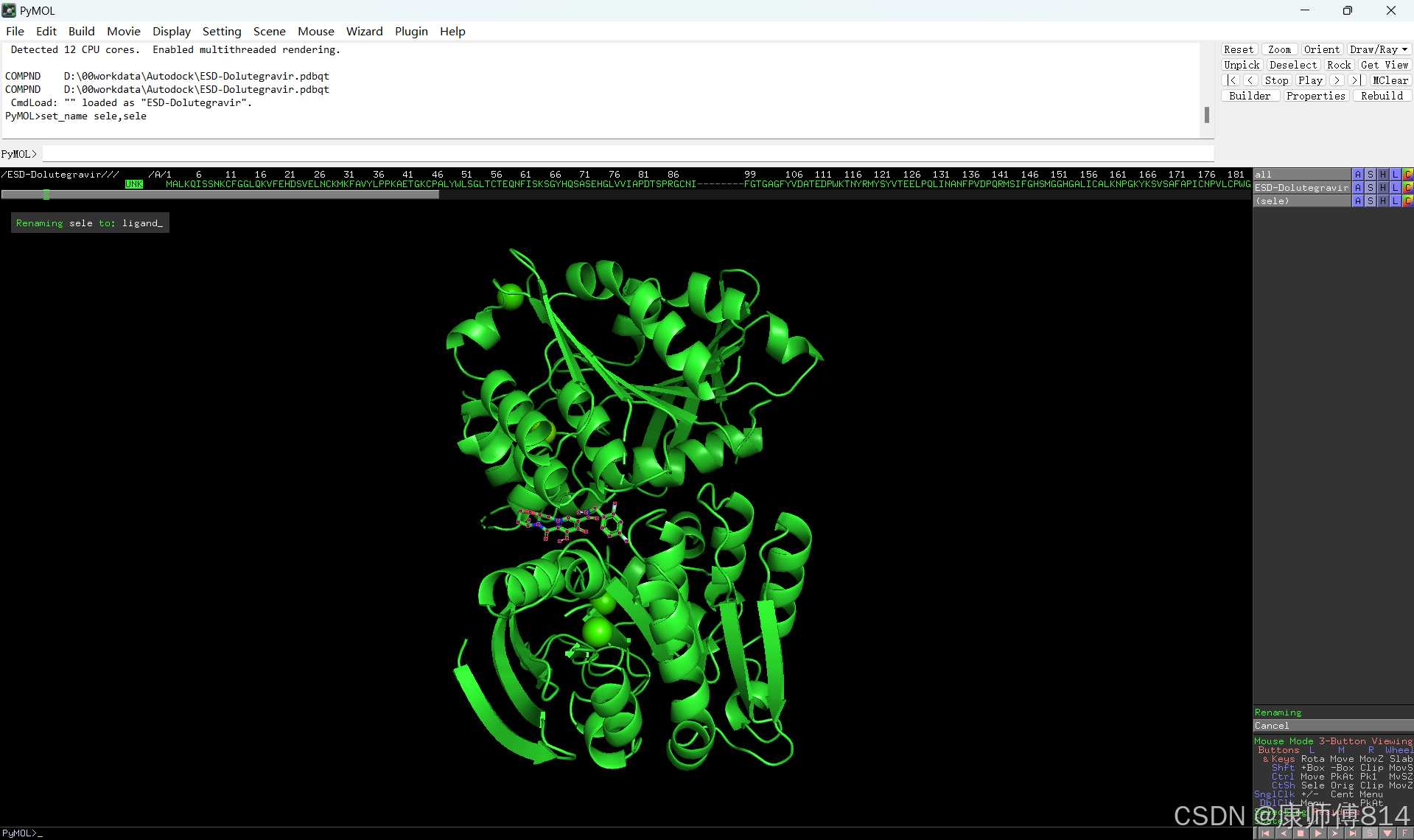
#在pymol中打开刚才得到的pdb文件,点击右下角S打开序列,选中小分子序列,命名为ligand。
#同样选中蛋白质序列,先将右下角Selecing选为Chains,然后才能悬着呢全部蛋白质序列(如果是多聚体蛋白,按住shift以连续选中多条链),命名为protein
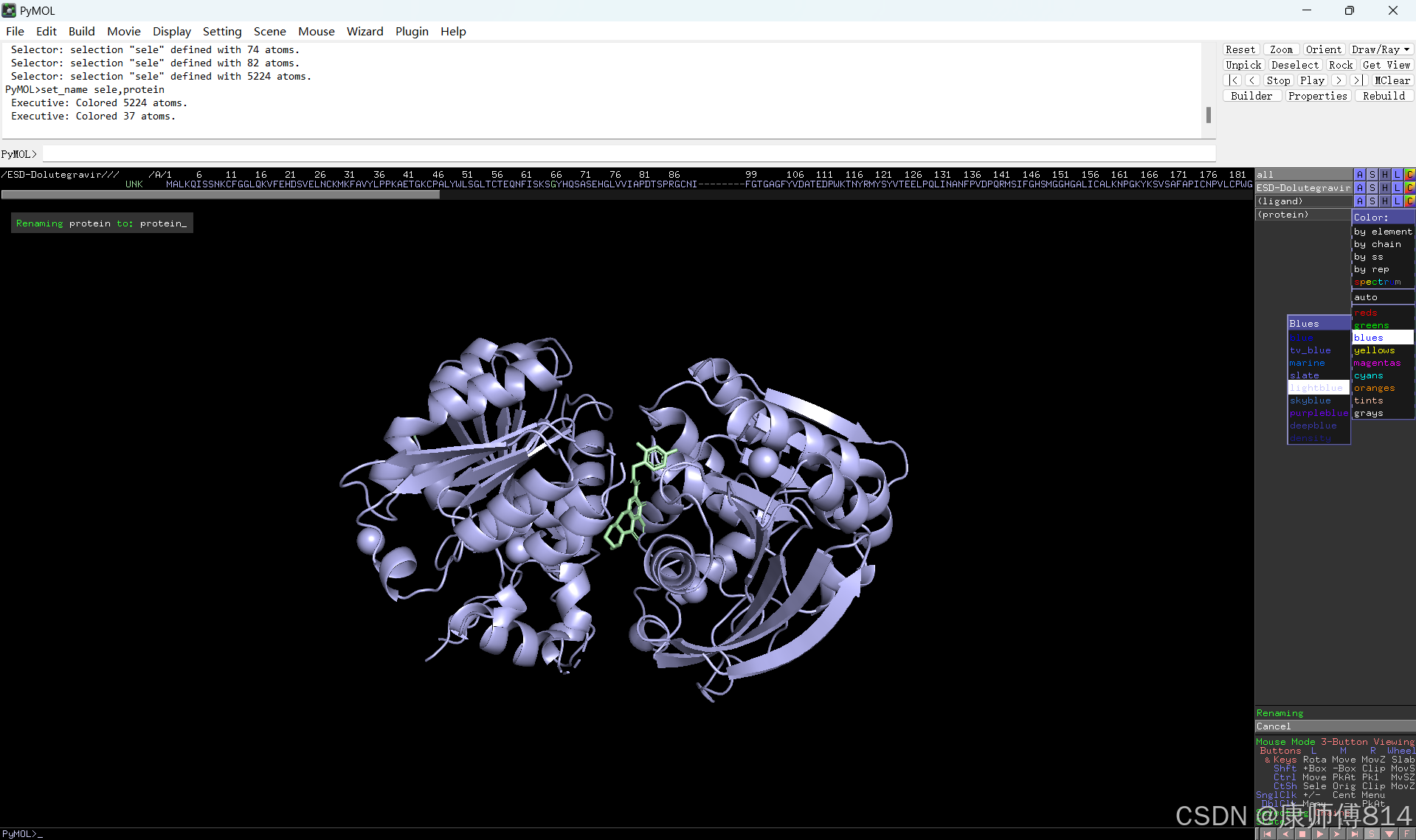
#点击蛋白和小分子后的C按钮可以修改颜色
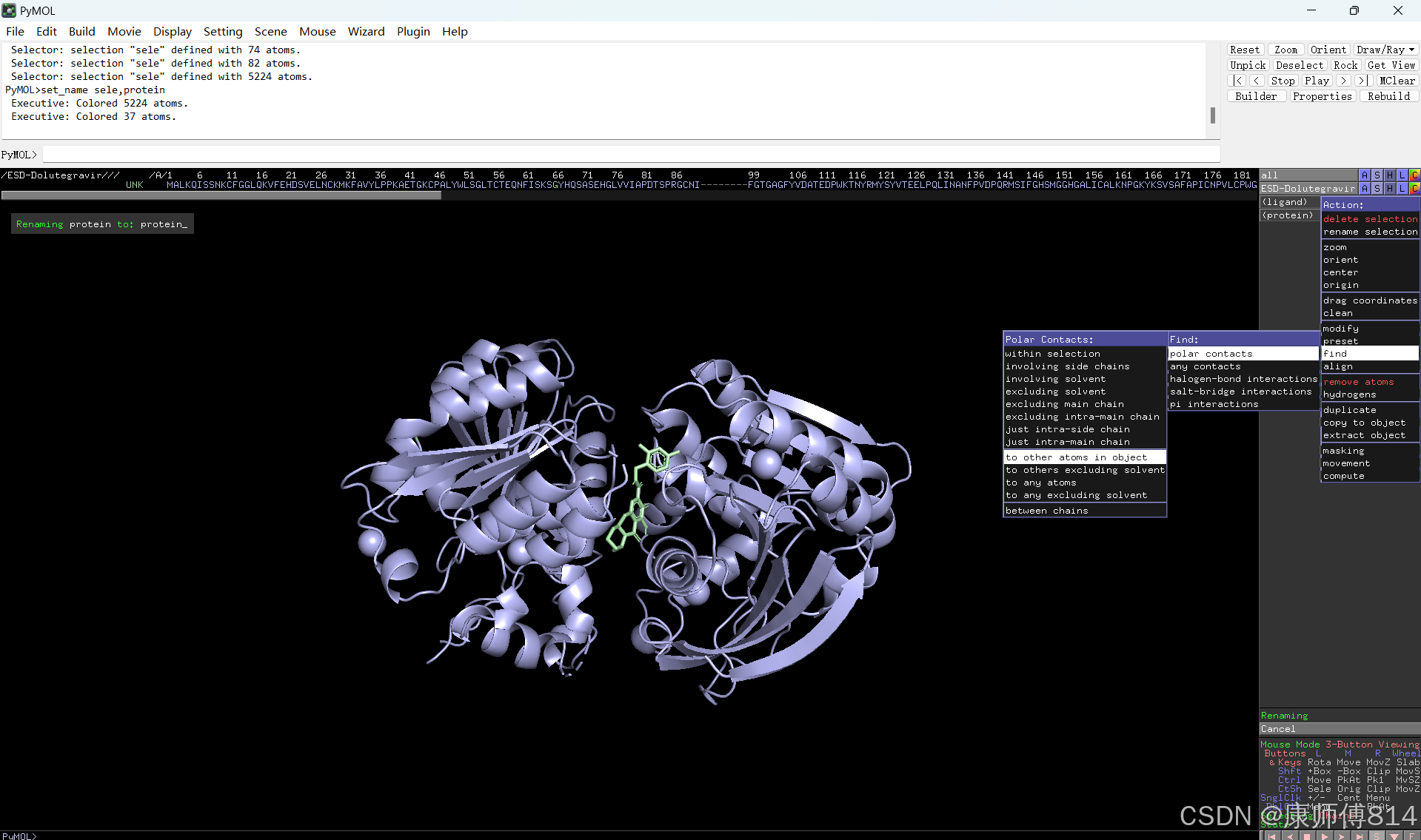
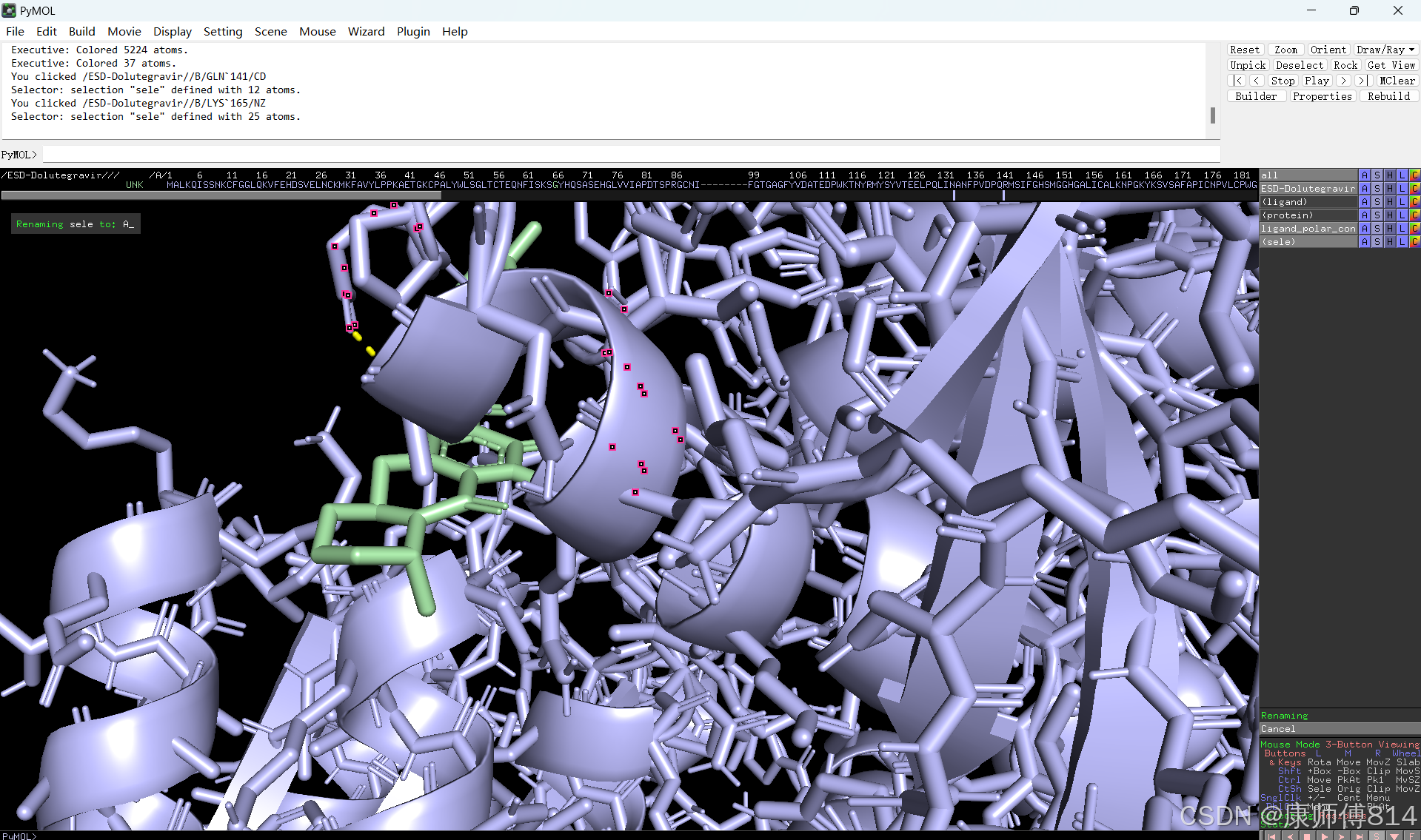
#选中小分子,按以下顺序点击以显示结合位点的氢键
#选中蛋白,点击show-sticks显示氨基酸棍状结构,右下角Selecting修改为Residues模式,选中与小分子连接的氨基酸残基,命名为A,并更改颜色
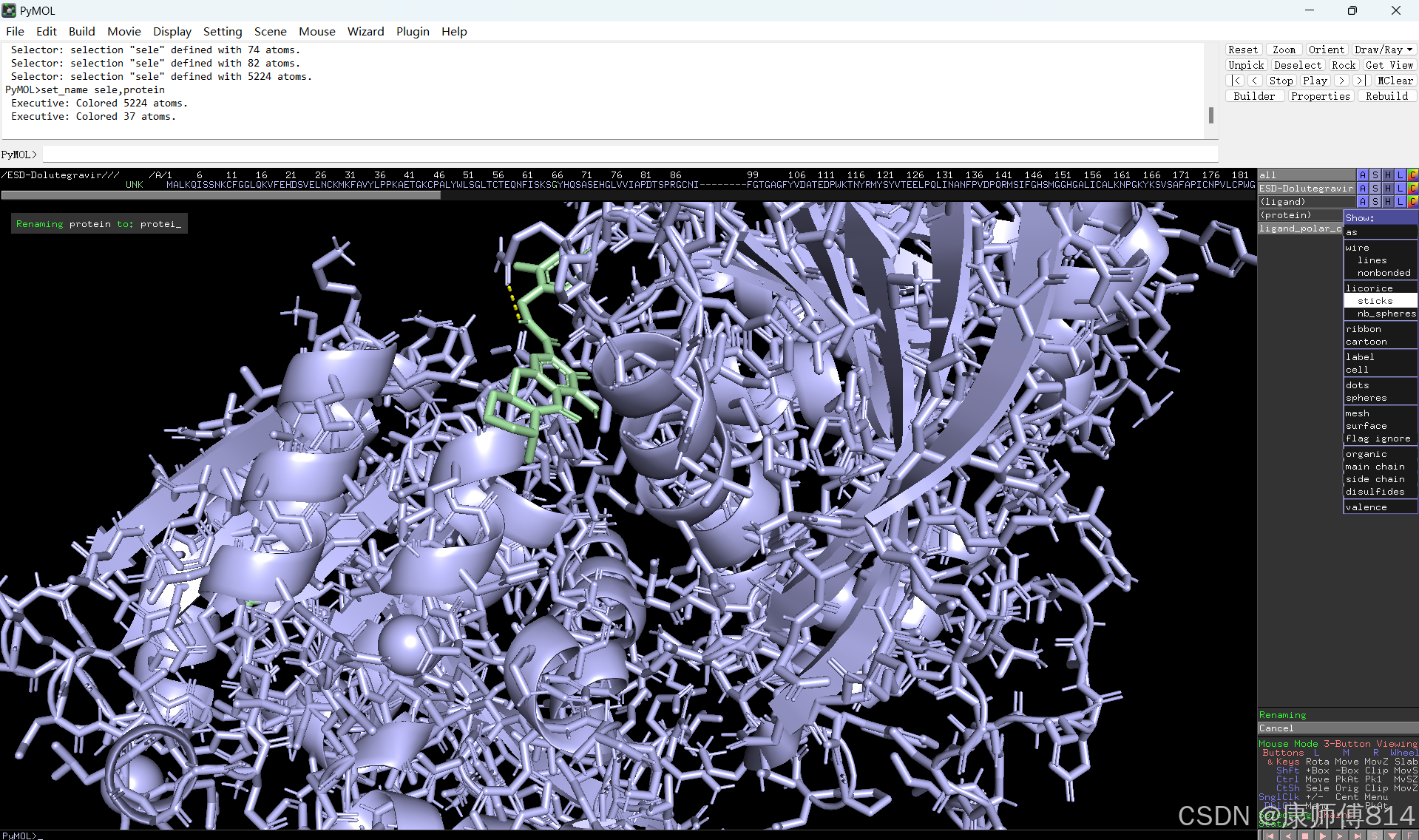
#点击蛋白,点击H-sicks隐藏蛋白棍状结构,然后显示小分子结合位点氨基酸残基棍状结构
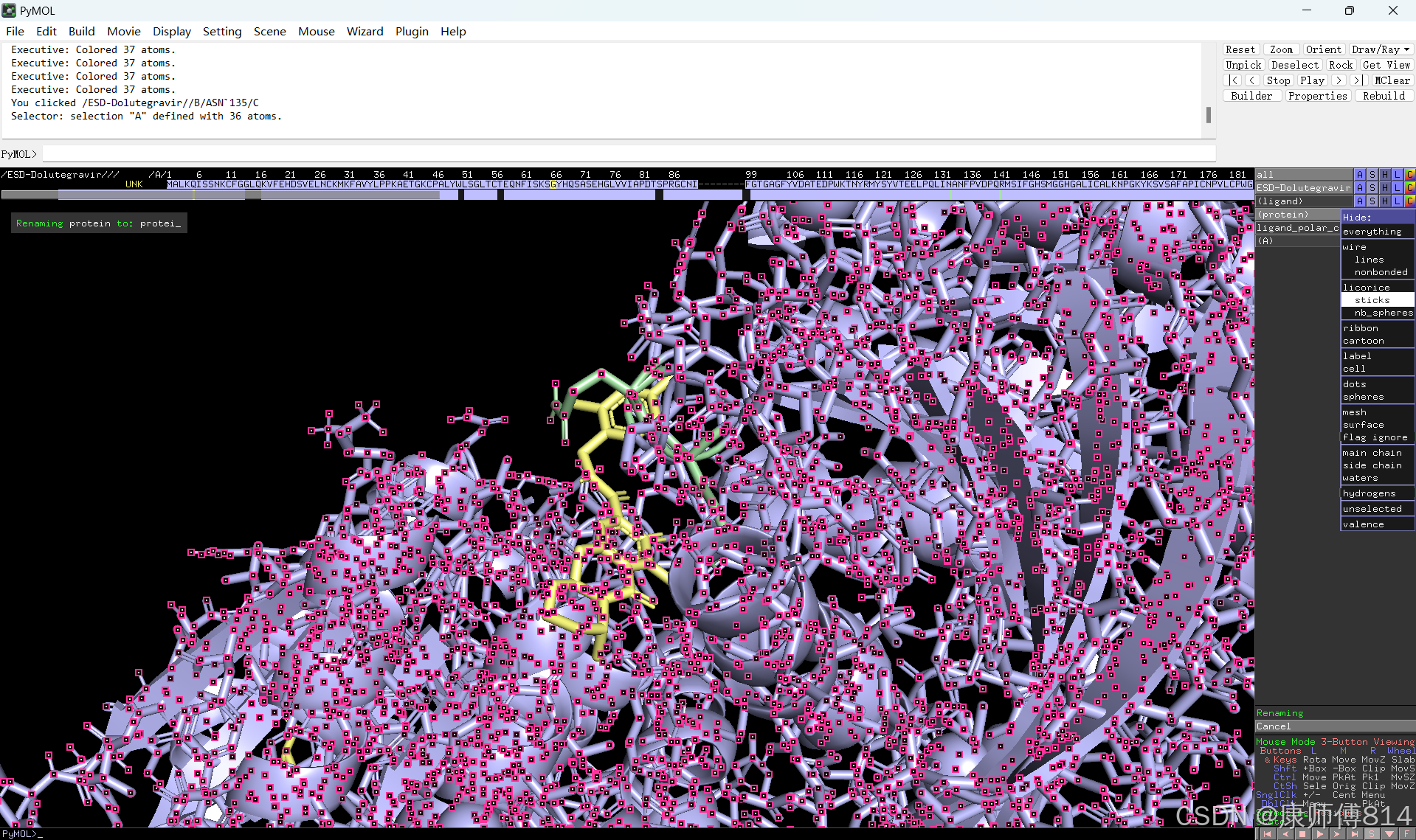
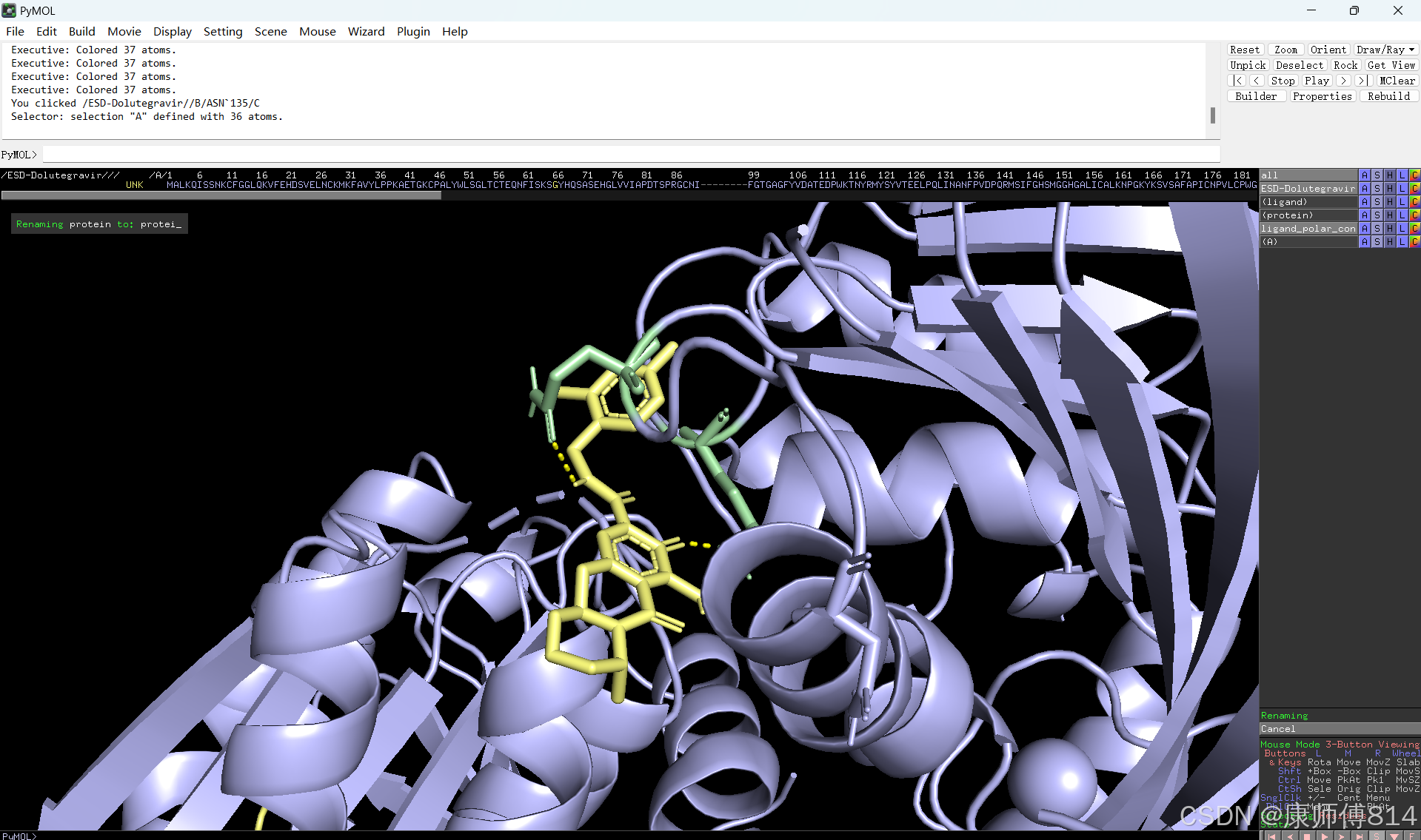
#此时展示分子对接的基本要素已经形成,后续可根据需要修改背景颜色、显示氨基酸残基名字以及氢键键能等等,这里不再赘述

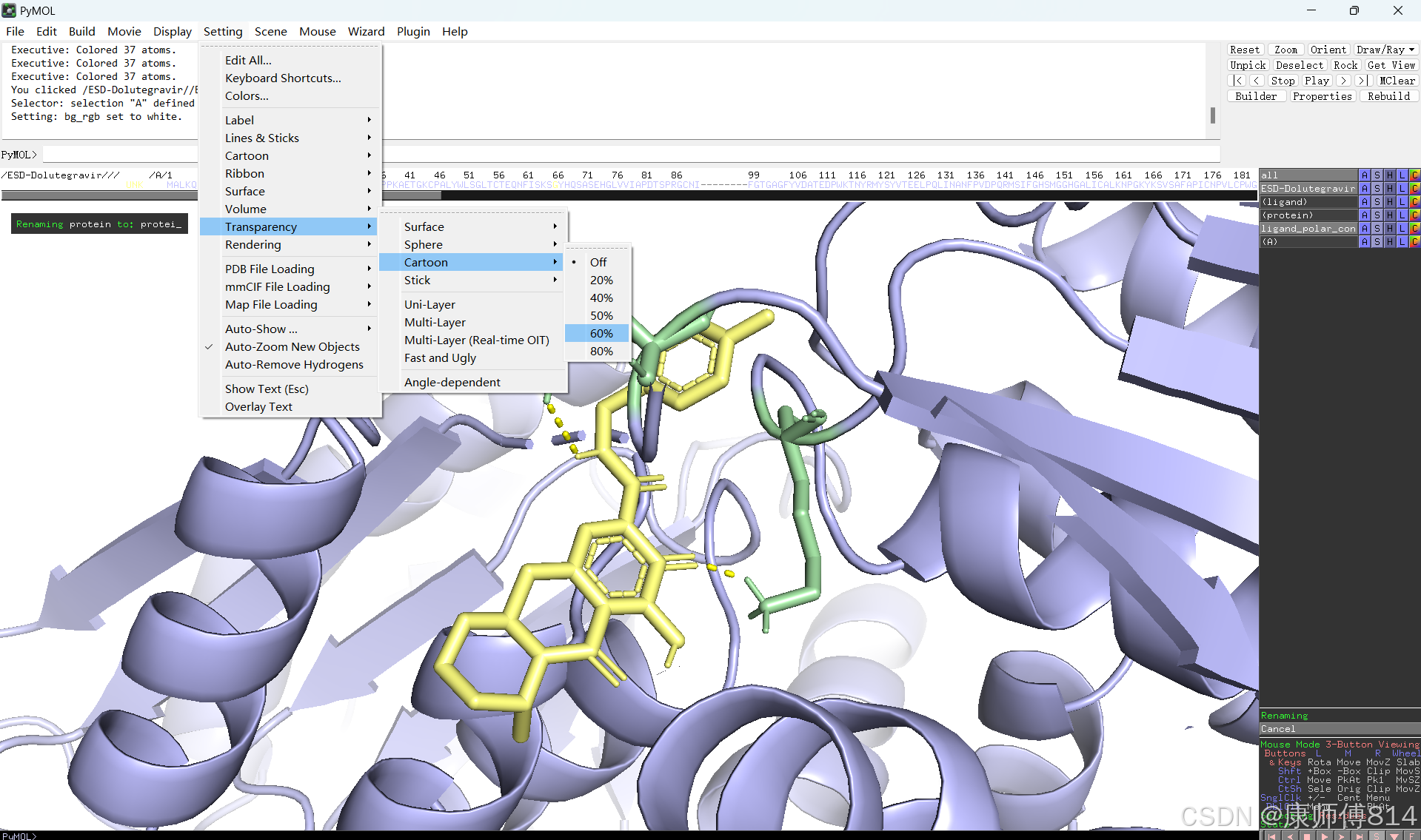
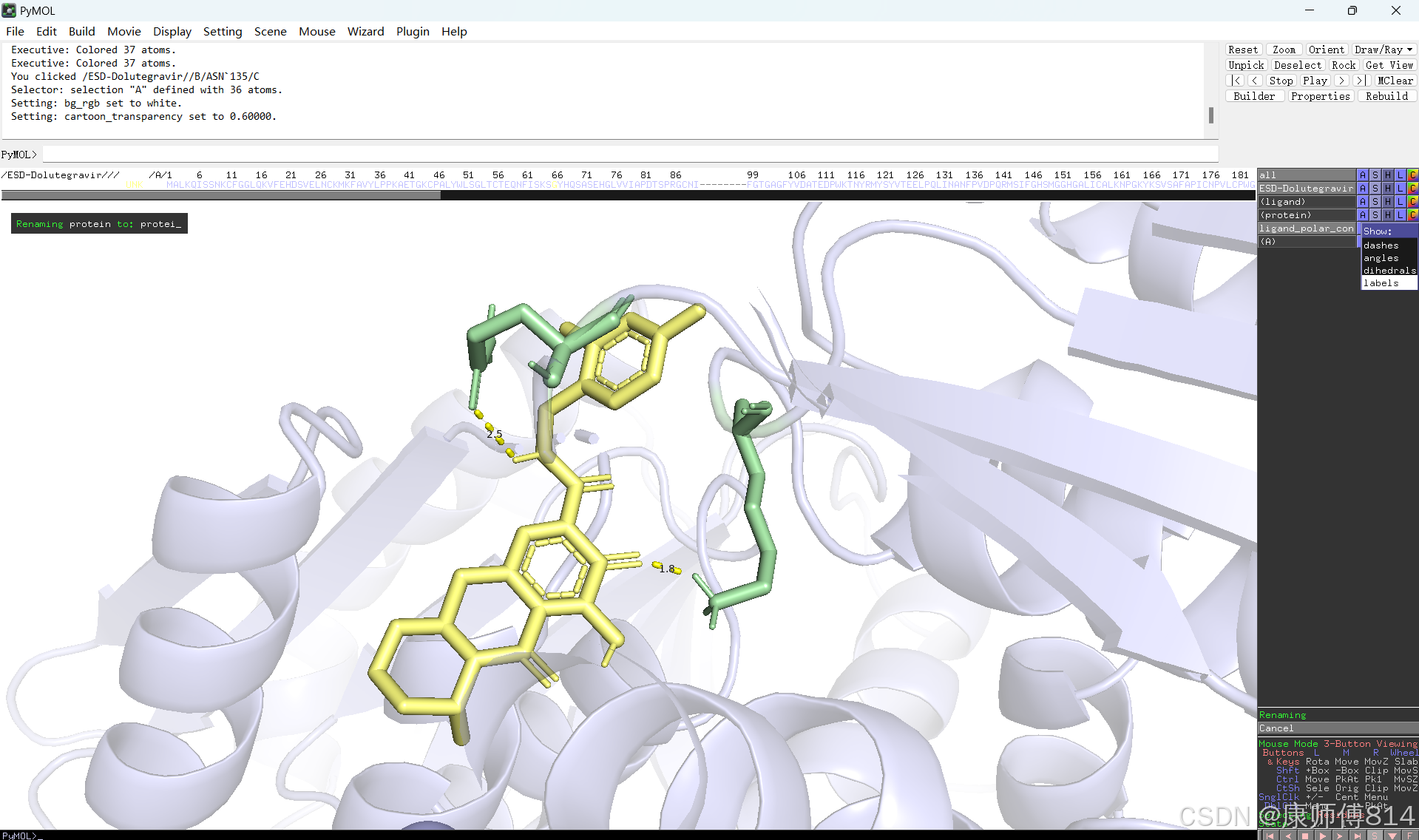
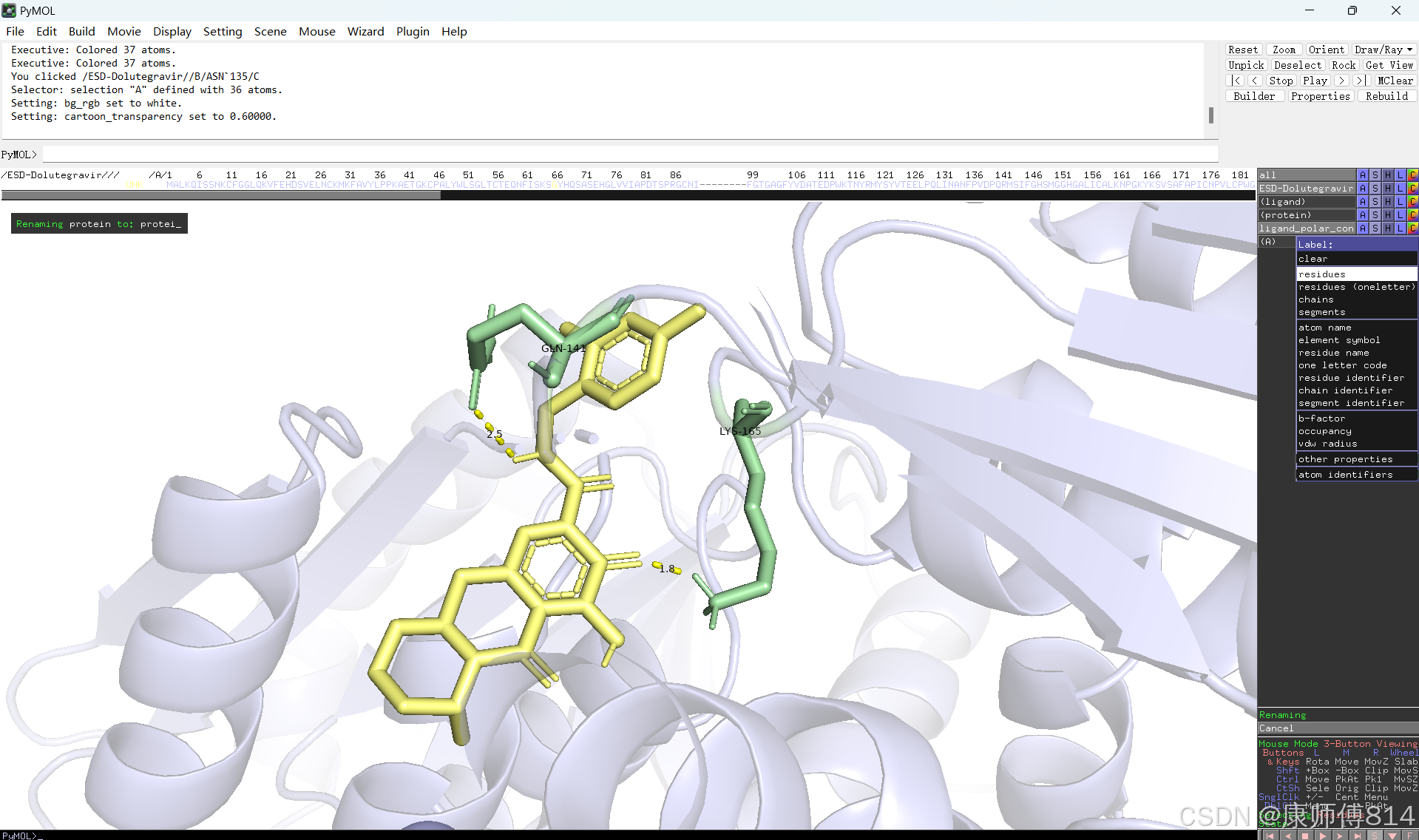
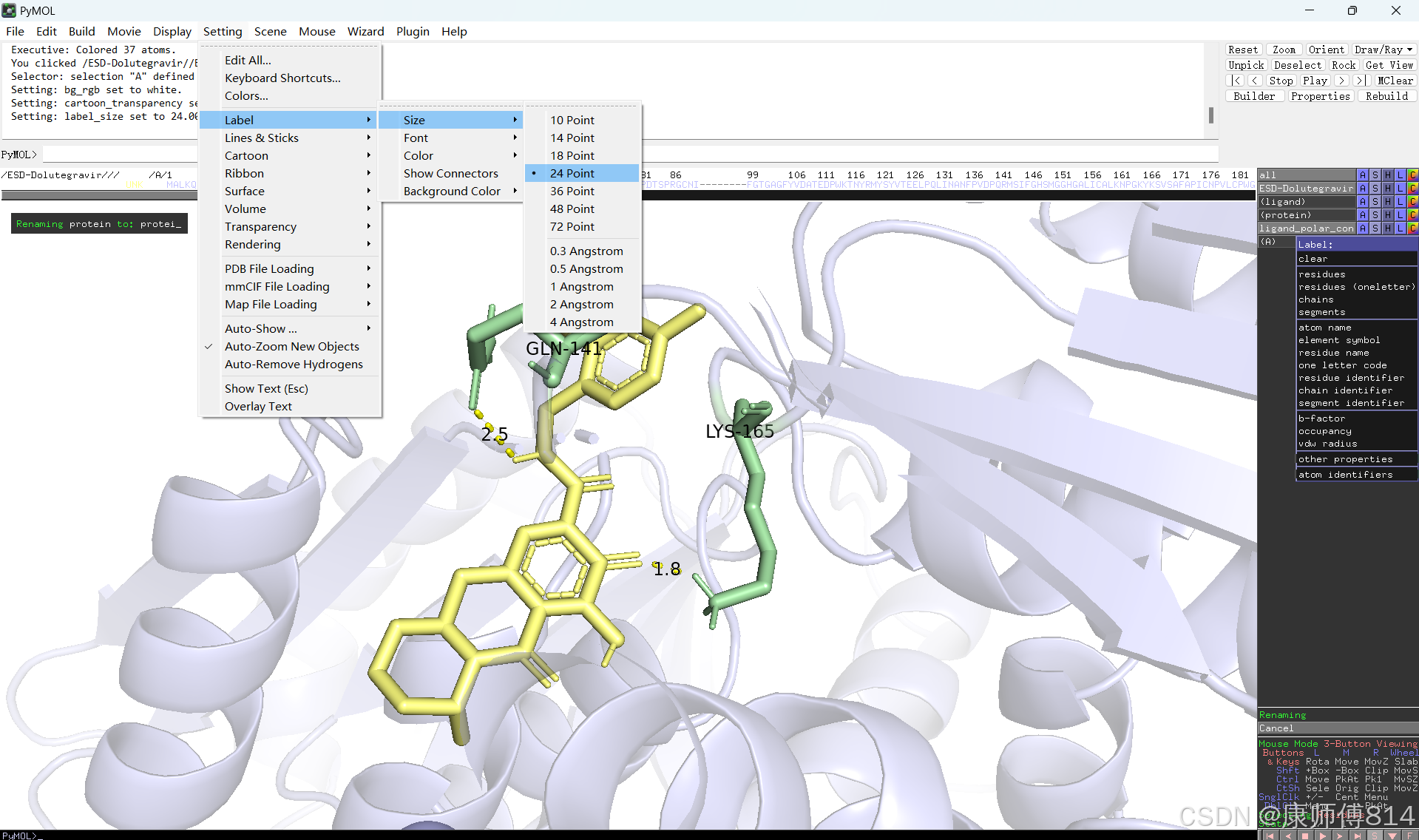
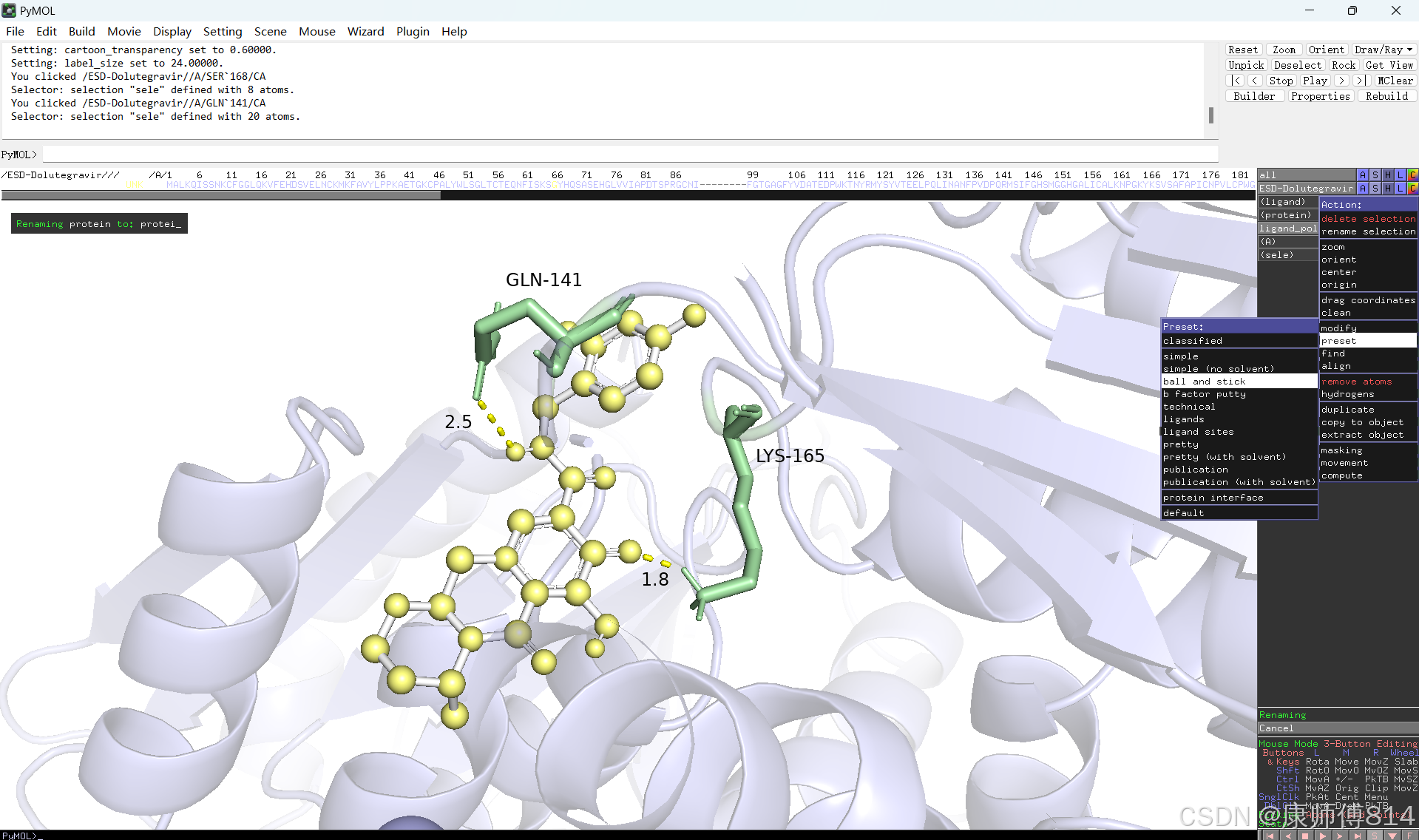
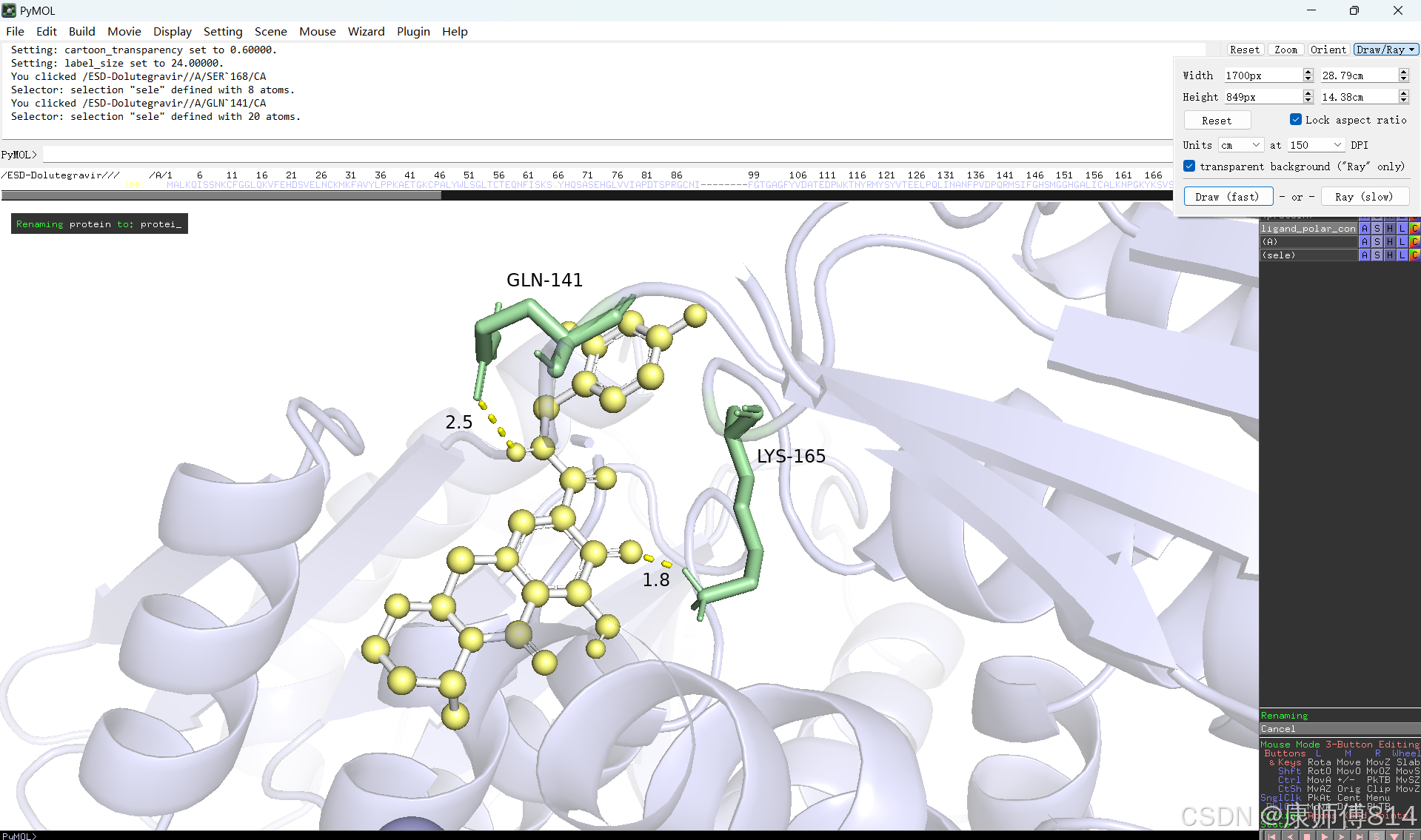
#修改好后就可以导出了,点击右上角Draw,选择合适的格式导出即可
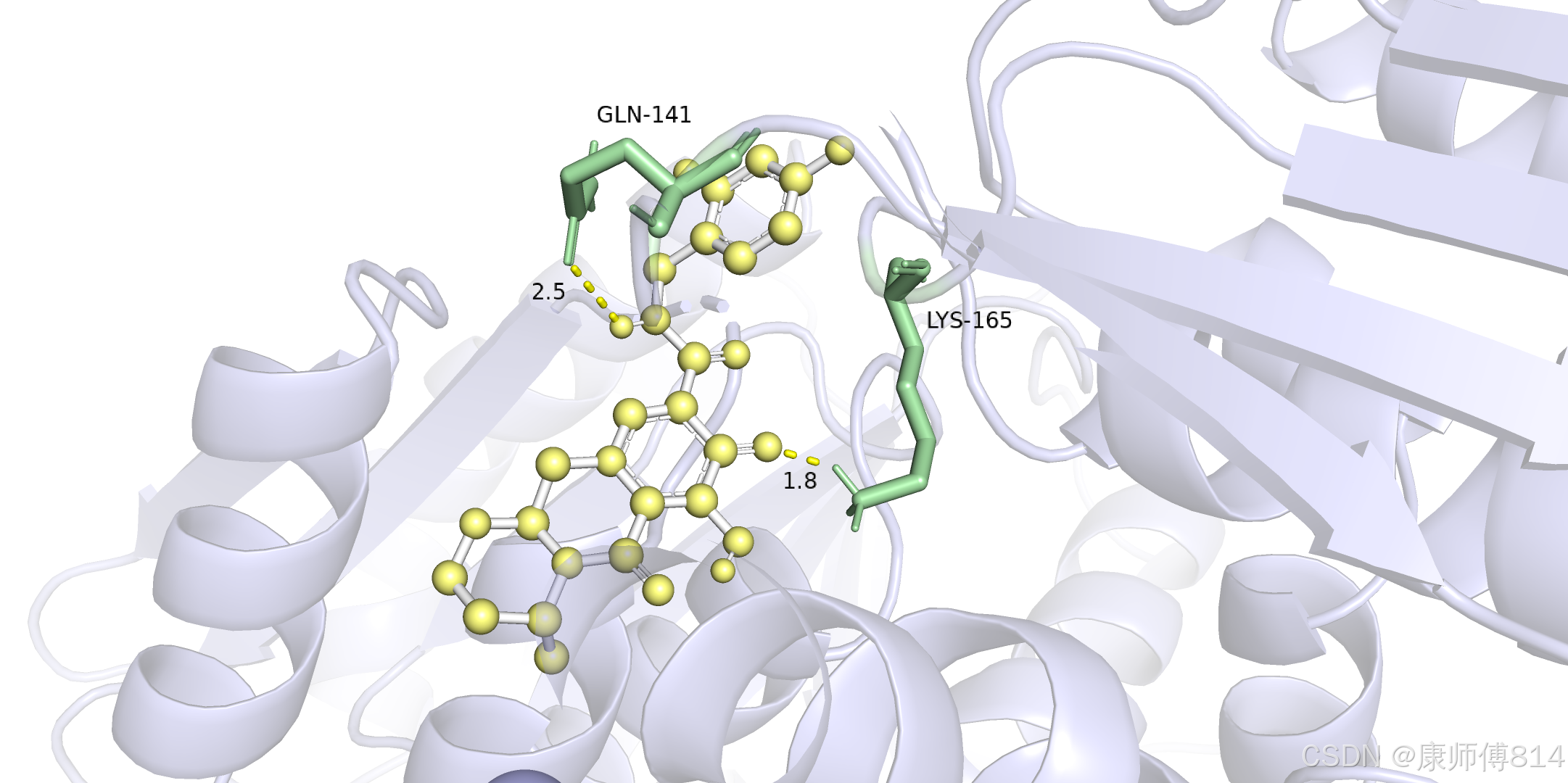
结果演示
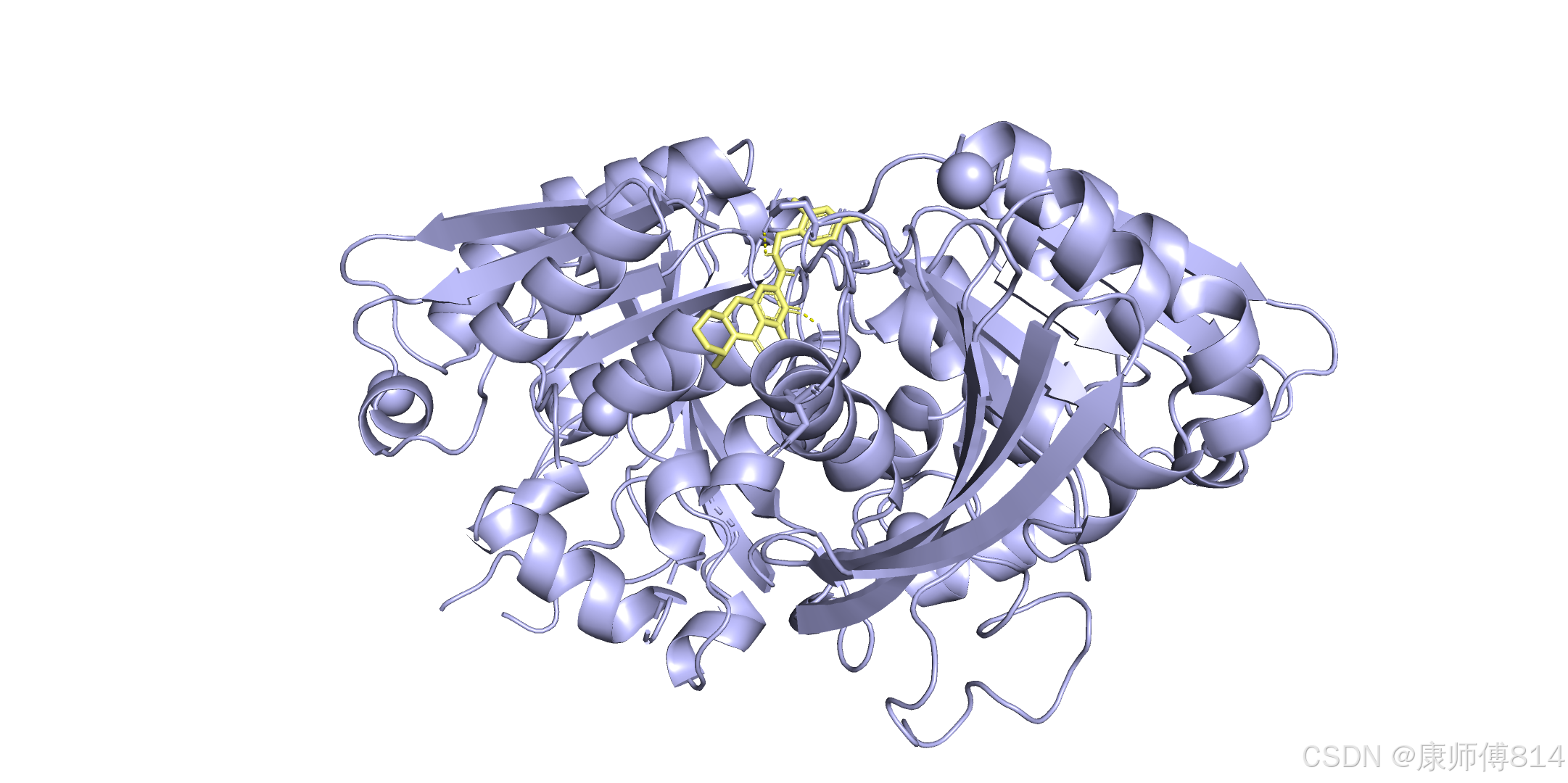
参考链接
https://www.bilibili.com/video/BV1Yr4y1T7Ko
https://www.bilibili.com/video/BV1M94y1k7aL

















 1957
1957

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









