







摘要
训练数据:860亿个氨基酸(来自2.5亿条蛋白质序列中的进化多样性
训练方式:无监督训练
成果:information about biological properties(有关生物特性的信息。
学习到的表示空间具有多尺度的组织结构,反映了从氨基酸的生化特性水平到蛋白质的远端同源性的结构。关于二级和三级结构的信息被编码在表示法中,并且可以通过线性投影来识别。
什么是氨基酸的生化特性水平?
什么是蛋白质的远端同源性的结构?
什么是t-SNE?
t-SNE (t-distributed stochastic neighbor embedding) 是一种非线性降维技术,用于将高维数据映射到低维空间,以便进行可视化和数据分析。它主要用于探索高维数据集的内在结构,尤其是在聚类分析和数据可视化方面非常有用。
为了验证跨领域后 the models and objective functions effective,作者训练了 high-capacity Transformer language models on evolutionary data
什么是evolutionary data?
进化数据指的是从各种生物体中收集的大量蛋白质序列数据,这些数据跨越进化历史。这些数据被用于研究蛋白质序列随时间变化和变异的情况,反映了进化过程。
进化数据具有几个重要用途,包括:
利用人工智能研究生物学的预测性和生成模型。 研究不同层次上的蛋白质结构和功能,如氨基酸、蛋白质和进化同源。 了解单个蛋白质的突变适应性景观,从而洞察序列与蛋白质功能之间的定量关系。 为在人工智能和生物学领域的表示学习和生成建模训练和评估高容量神经网络架构。
MSA(multiple sequence alignment)是什么?
是生物信息学中常用的一种技术,用于比较和分析多个生物序列(如蛋白质或核酸序列)之间的相似性和差异性。通过多序列比对,可以揭示不同序列之间的保守区域、变异区域以及特定结构或功能相关的区域。
多序列比对通常包括以下步骤:
- 选择要比对的多个序列,这些序列可能来自不同物种、同一物种的不同个体或同一基因的不同亚型。
- 使用比对算法(如ClustalW、MAFFT、T-Coffee等)对这些序列进行比对,将它们在序列上的相似部分对齐。
- 分析比对结果,揭示序列之间的保守区域和变异区域,推断共同祖先序列以及可能的结构或功能相关区域。
- 根据比对结果进行进一步的生物信息学分析,如构建系统发育树、预测蛋白质结构、识别功能位点等。
多序列比对在研究基因组学、蛋白质结构与功能、系统发育学等领域都具有重要意义,帮助科学家理解生物序列之间的关系及其在进化和生物功能中的作用。
简介
我们在这里的重点将是将一个单一的模型适合于跨越进化的许多不同的序列。
在蛋白质序列中可能存在各种随机的扰动,但进化倾向于选择那些与生物适应性(fitness)一致的扰动(evolution is biased toward selecting those that are consistent with fitness)。换句话说,生物在进化过程中更可能选择对其生存和繁殖有利的蛋白质序列变异,这些变异使得生物更适应其所处的环境和生存条件。(适者生存:Survival of the fittest)
贡献1:We find metric structure in the representation space that accords with organizing principles at scales from physicochemical to remote homology.
翻译:我们发现representation空间中的度量结构符合从物理化学到遥远同源性等尺度的组织原则。
贡献2:We also find that secondary and tertiary protein structure can be identified in representations. The structural properties captured by the representations generalize across folds.
翻译:我们还发现,二级和三级蛋白质结构可以在representations中识别出来。表征所捕捉到的结构特性在不同fold之间具有普遍性。
fold是什么?
"fold" 是蛋白质结构领域中的一个术语,表示蛋白质的折叠形态或结构类型。蛋白质折叠是指蛋白质在其功能状态下的三维结构形态,这种形态由其氨基酸序列所决定。
蛋白质fold仅由氨基酸顺序决定吗?
一级序列中氨基酸的疏水效应、氢键决定了二级结构α-螺旋(α-helix)、β-折叠(β-sheet)、β-转角(β-turn)Ω-环(Ω-loop)等,二级结构进一步可以形成超二级结构/模体,在此之上还有三级结构(疏水效应、二硫键、离子键、氢键)、四级结构(疏水效应、离子键、氢键维持),这些非化学键的作用都是由一级结构决定的,典型的就是二硫键,是一级结构中巯基之间形成的,离子键、疏水效应和氢键也都与氨基酸残基有关,因此可以认为一级结构决定了空间结构。
贡献3:We apply the representations to a range of prediction tasks and find that they improve state-of-art features across the applications.
翻译:我们将这些表征应用到一系列预测任务中,发现它们改善了各种应用中的最新特征。
训练
train models using the masked language modeling objective (Devlin et al., 2018)(BERT).

UniProt is a comprehensive and freely accessible database of protein sequence and functional information.
UniRef50, a clustering of UniParc(UniProt Archive) at 50% sequence identity.
cluster是什么?
"UniRef50 clusters" 指的是 UniRef50 数据集中的聚类,其中包含具有相似序列的蛋白质序列。
数据集:UR50/S
参数规模:650M
模型层数:33
成果
学习到了氨基酸生物特性(Learning encodes biochemical properties)
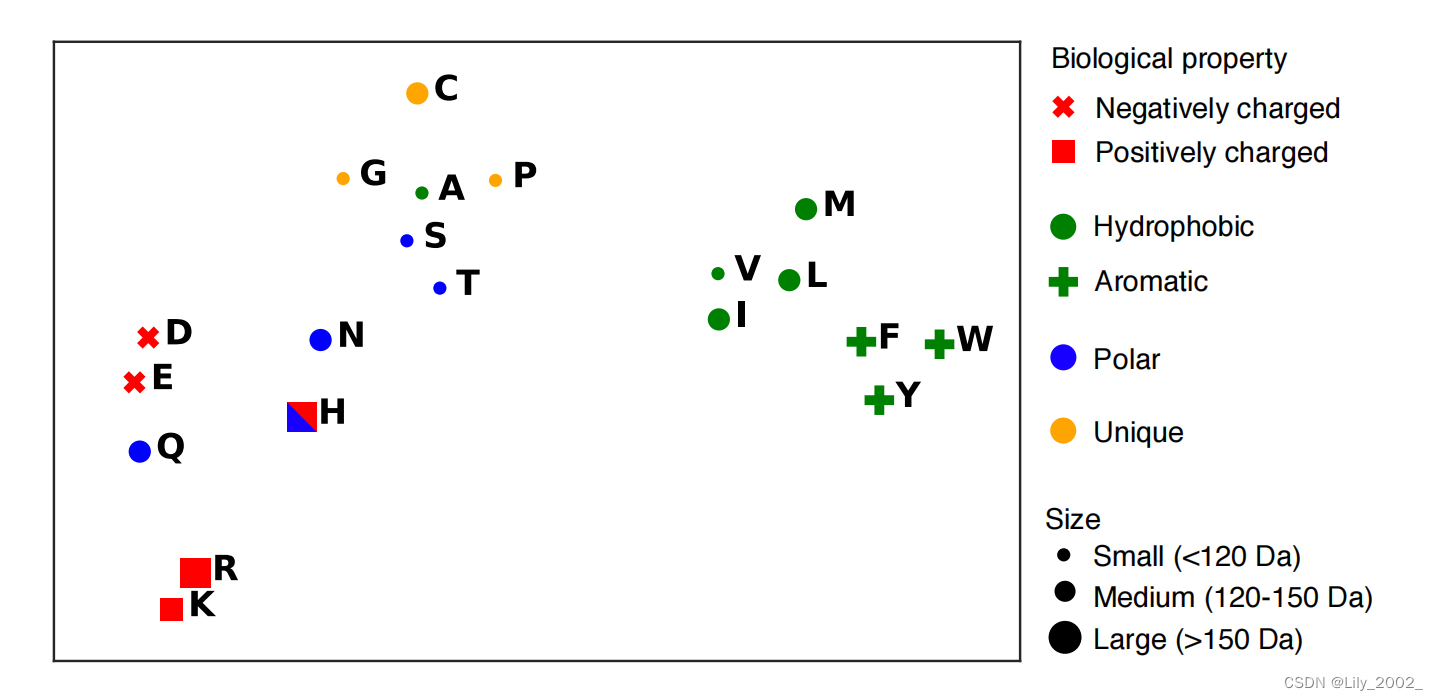
氨基酸的生化特性在Transformer模型的输出representation中表示,这里用t-SNE可视化。通过无监督学习,残基被聚为疏水、极性和芳香族基团,并通过分子量和电荷反映整体组织
生物变异(Biological variations are encoded in representation space)

(a)t-SNE: 颜色----orthologous group
经过训练后,直系同源组的基因都聚集在一起
(b)PCA: 横坐标----a species axis, 纵坐标----orthology axis
颜色----orthologous group, 字母----species
经过训练后,同源的在水平线上,同一物种在垂直线上
远缘同源(Learning encodes remote homology)
蛋白质家族中进行序列比对(Learning encodes alignment within a protein family)

未经训练时(红),Aligned pairs和Unaligned pairs区分不出,训练之后(b)即可区分。并且,不论residue pairs有没有相同的amino acid identities这种趋势会持续
下游任务

二级结构预测

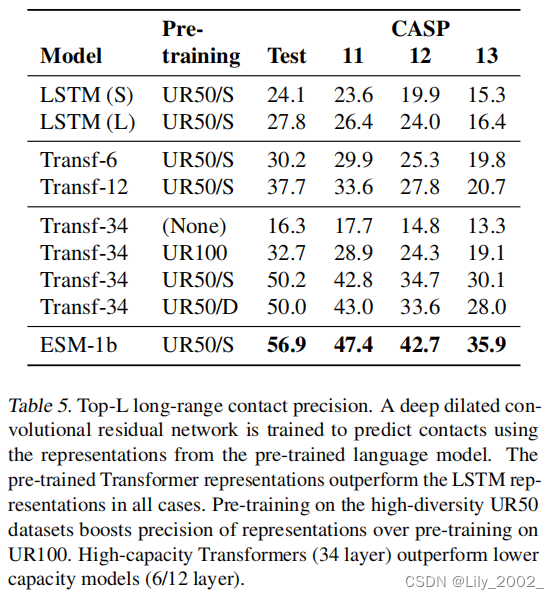
三级结构预测















 1630
1630











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









