









Problem Description:
The CG island is a stretch of DNA (usually longer than 200 bases) in which the frequency of the CG sequence is higher than other regions. It is also called the CpG island, where "p" simply indicates that "C" and "G" are connected by a phosphodiester bond.
CpG islands are often located around the promoters of housekeeping genes (which are essential for general cell functions) or other genes frequently expressed in a cell. At these locations, the CG sequence is not methylated. By contrast, the CG sequences in inactive genes are usually methylated to suppress their expression. The methylated cytosine may be converted to thymine by accidental deamination. Unlike the cytosine to uracil mutation which is efficiently repaired, the cytosine to thymine mutation can be corrected only by the mismatch repair which is very inefficient. Hence, over evolutionary time scales, the methylated CG sequence will be converted to the TG sequence.
This explains the deficiency of the CG sequence in inactive genes.
The following example was taken from Chromosome #1 in the Human Genome. Notice the higher concentration of G+C in the CpG island near Base #2000. In this case, the minimum length was set to be 200 nucleotides, so the second peak around Base #5200 was not recognized
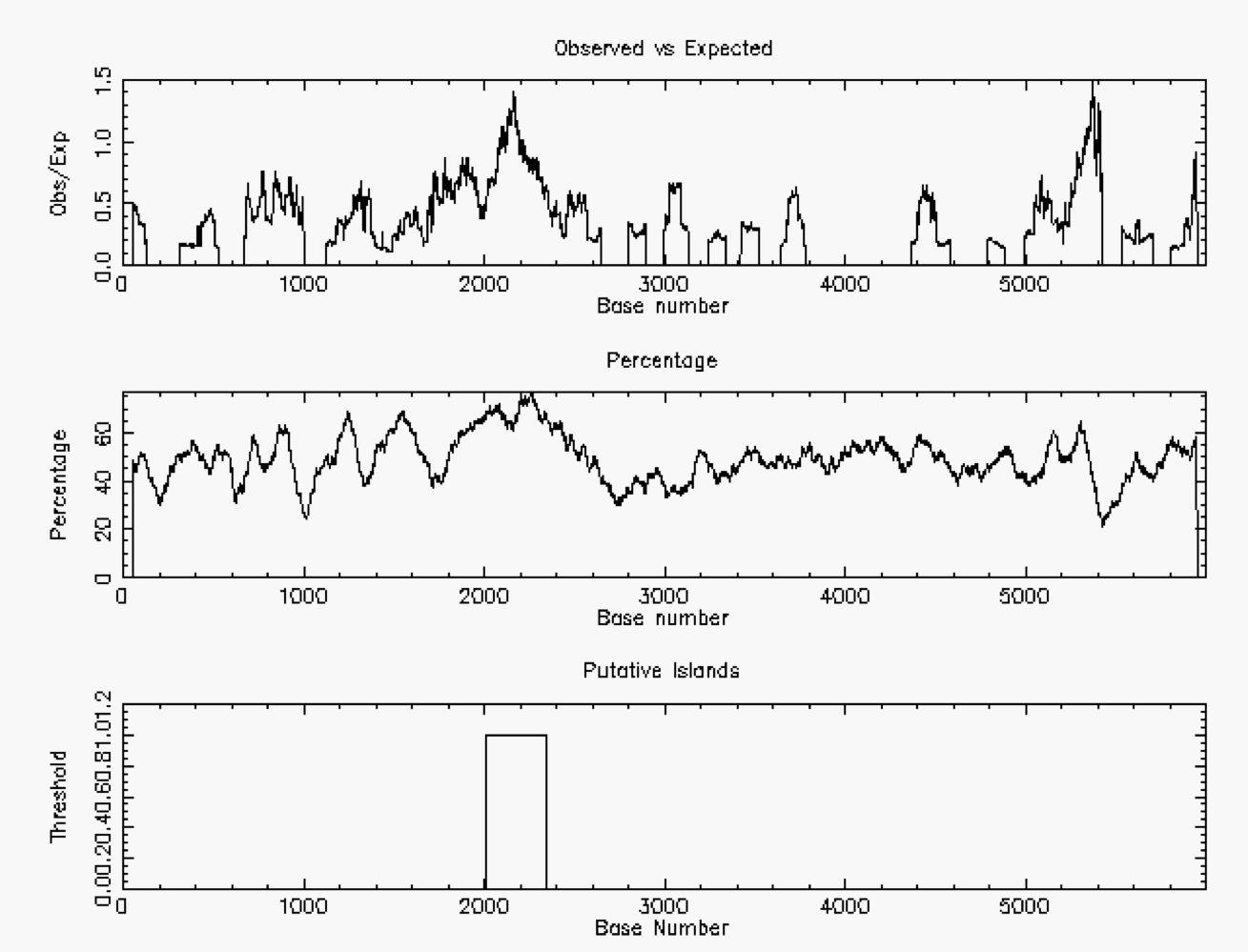
CpG islands occur near the beginning of a gene in the human genome and can be used for gene finding. In these Islands, there will be a more equal concentration of c and g nucleotides. In inactive regions, there will be a higher concentration of t nucleotides than c nucleotides.
Assignment:
In this assignment, you will write code for a Hidden Markov Model to find CpG islands. You will implement the Viterbi algorithm to find the most probable sequence of hidden states for the following model. Then you will use annotated data to find model parameters for your HMM and evaluate your accuracy.
Background:
We will use the following general definition of a Hidden Markov Model for this description.

It is easy to see how you can generate the probability of a given sequence of hidden states, using this construct.
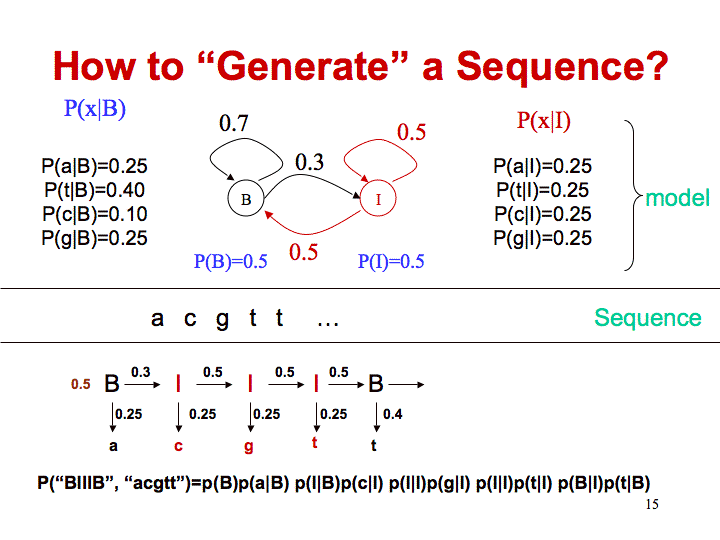
Find the island using Viterbi
In finding a CPG island, we would like to determine the most likely path through the model.
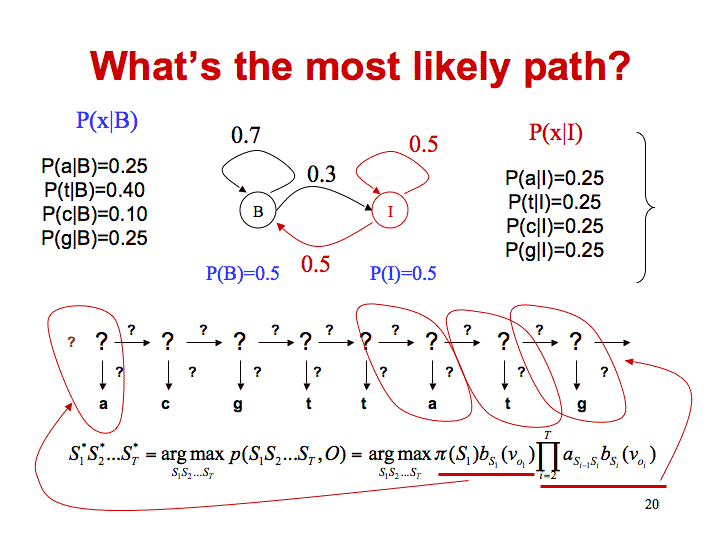
There are two ways of doing this. If you are interested in the overall probability of a sequence of states, then you use the forward algorithm which requires you to sum the probabilities of all possible previous states. The Viterbi algorithm just uses the previous state that generates the most probable arrival at the current state and takes the product of these max probabilities to select the correct path. The following example will illustrate the algorithm.
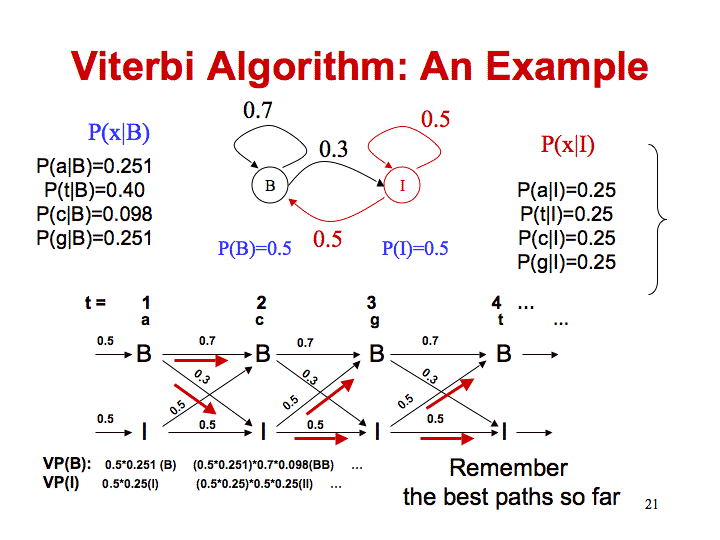
In this example, both states have a 0.5 probability, so the probability of being in state B is (0.5 * 0.251) with 0.251 being the probability of emitting an a (this is the observed first character) while in state B.
The probability of being in state I for the initial state is (0.5 * 0.25) with 0.25 being the probability of emitting an a (this is the observed first character) while in state I.
In order to calculate the probability of being in state B at time=2, Viterbi calculates the max probability of coming from states I or B in the previous state.
- Coming from state B we use the probability of being in state B at time=1 (0.5 * 0.251). Multiply by the probability of staying in state B (0.7). The total in this case is 0.5*0.251*0.7.
- Coming from state I we use the probability of being in state I at time=1 (0.5 * 0.25). Multiply by the probability of moving from state I to state B (0.5). The total in this case is 0.5*0.25*0.5.
Since the probability is higher for coming from state B, we will use this to calculate the probability of being in state B at time=2. Since the probability of emitting a c in state B is 0.098, the total probability of being in state B at time=2 is (0.5*0.251*0.7)*0.098.
In order to calculate the probability of being in state I at time=2, Viterbi calculates the max probability of coming from states I or B in the previous state.
- Coming from state B we use the probability of being in state B at time=1 (0.5 * 0.251). Multiply by the probability of moving from state B to I (0.3). The total in this case is 0.5*0.251*0.3.
- Coming from state I we use the probability of being in state I at time=1 (0.5 * 0.25). Multiply by the probability of staying in state I (0.5). The total in this case is 0.5*0.25*0.5.
Since the probability is higher for coming from state I, we will use this to calculate the probability of being in state I at time=2. Since the probability of emitting a c in state I is 0.25, the total probability of being in state B at time=2 is (0.5*0.25*0.5)*0.25.
This same procedure can be used to calculate the probabilities of being in each state until the whole sequence has been analyzed. You can compute the log likelihood by using the logarithm of the probabilities instead of the actual values. You then substitute addition for multiplication. Since the probability of the state at the previous time step is already in log form, you just add it, instead of taking the log of it.
You should experiment with different values for emission and state transition probabilities to determine values that categorize the data most accurately.
In order to find an island, slide a window across your most probable state list looking for a threshold of Island states. You will have to determine the threshold values. Plot the number of I states in the window and turn it in as part of your writeup.
Estimating Model Parameters
You will probably find that the model parameters given are not very good at discriminating between islands and normal DNA. The next part of the assignment will give you the opportunity to calculate model parameters. Although sophisticated algorithms can be used in unsupervised training, we are going to use a very simple approach.
Using annotated sequence data, calculate the Bayesian probability of each emission or transition. For example, if you observe that when you are in state B, you transition to state I 100 times and stay in state B 200 times, then the probability of B->A transitions should be 0.33 and the probability of B->B should be 0.66. Count up the emissions from each state. If the totals from state B are a->20, c->30, t->40, g->60, then the probability of emitting a t in state B is 40/(60+40+30+20). Use this model based on supervised training to find islands and compare it to your initial results. You can use this annotated data along with that you find in other places to train your model.

Deliverables
- A description of your HMM implementation and any problems you had in writing the code using the Lab Report Guidelines.
- Your analysis of this sequence data or fasta sequence data from Human Chromosome 1 using your HMM. Plot the percentage of I states over the sequence data.
- You should train your parameters using existing data
- You should run your trained model on other annotated data that you collect from genbank.
- You can get 30% extra credit for doing Baum-Welsh
Hints
- You may want to look at this spreadsheet to validate your algorithm.
- You can't determine the state for a particular time slot by just looking at the probability. You have to start from the end, pick the most probably state, then decide which previous state was used in the maximization, and so forth.
- The first set of rows use the normal probabilities (no logs)
- The second group use the log computations. I also included the raw greater than state predictions (we mentioned that these will be wrong) so that you could compare them with the correct computations.
- The third group show the unlogged values to give you confidence that you get back the same results you would have had if you used normal (non-log) probabilities.
- The fourth group predicts states using the traceback algorithm. It breaks the computation up so it is easier to do on the spreadsheet. For each time slot, it calculates the probability of getting to the state from each of the previous time slot states and then uses this to start from the max time slot and compute previous values. Let me know if you see problems.
- You might want to work through this ghmm tutorial.
- You can check your results with other programs that find cpg islands:
- cpgislands from USC provides a graphical view of the island if you use Internet Explorer. Here are the results for the Human Chromosome 1 data.
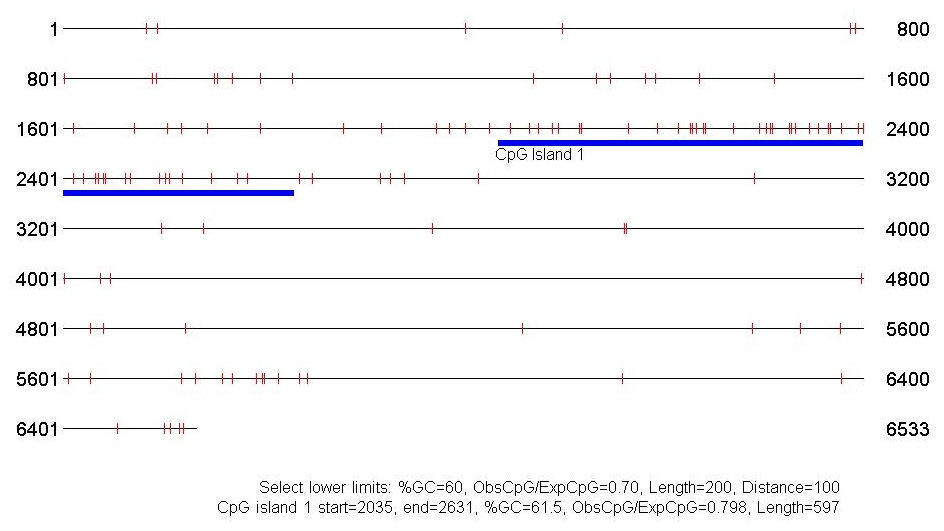
- The CpGProD application finds the following cpg islands. This application is also available in /usr/local/bin on psoda4.
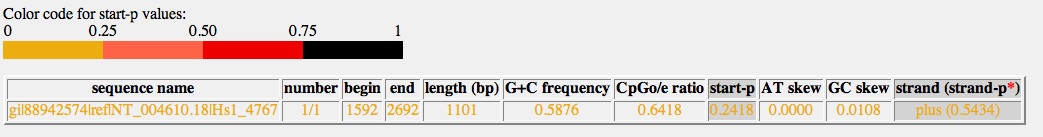
- The EMBOSS CpGPlot application finds the following cpg islands.
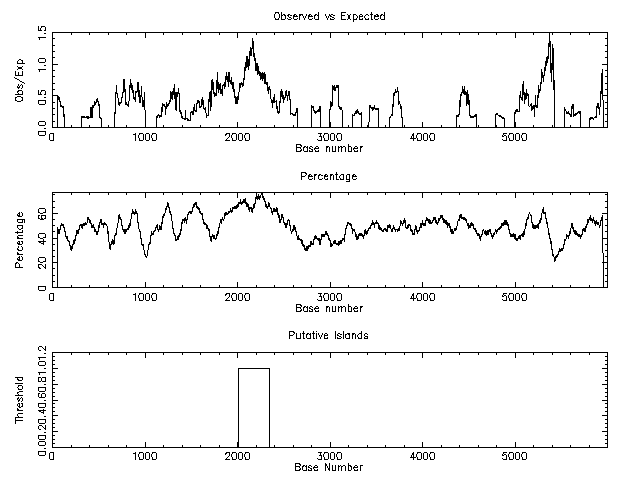
- cpgislands from USC provides a graphical view of the island if you use Internet Explorer. Here are the results for the Human Chromosome 1 data.
- You may want to look at this web site for added gene finding information.
- You may want to plot the percentage of C&G (%C+%G) in a sliding window of length > 200 in order to visualize where your predicted island is.
- If you are doing Baum-Welch training, you will have to deal with adding logorithms. This Tutorial on HMMs will give you some good tips on how to do this. This procedure raises a very important implementation issue. As a matter of fact, the computation of the alpha_t vector consists in products of a large number of values that are less than 1 (in general, significantly less than 1). Hence, after a few observations (t approx= 0), the values of alpha_t head exponentially to 0, and the oating point arithmetic precision is exceeded (even in the case of double precision arithmetics). Two solutions exist for that problem. One consists in scaling the values and undo the scaling at the end of the procedure : see [RJ93] for more explanations. The other solution consists in using log-likelihoods and log-probabilities, and to compute log p(X | theta) instead of p(X | theta).
log(a + b) = f(log a, log b) = log a + log {1 + e ^ (log b - log a) }-
来源:http://dna.cs.byu.edu/bio465/Labs/hmm.shtml
参考:http://en.wikipedia.org/wiki/Hidden_Markov_model

















 431
431

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









