







3.6 最优化分隔方案和最优氨基酸进化模型的选择
连接超级矩阵后,使用PartitionFinder 软件对相关矩阵进行分隔(分区)方案分析。输入初始定义的超级矩阵数据块,选择校正后的赤池信息量准则(Akaike information criterion,AICc)为数据块设定分区。(该标准权衡了更复杂模型改进的似然比得分与引入额外模型参数之间的关系。)
分区分析的原因:多个基因的变异速率存在差异,同一蛋白编码基因中编码同一氨基酸的三个位点的核苷酸的变异速率也存在差异,数据进行分区,分别计算不同区的最优模型,才能对这些位点设置其最优的核苷酸替换模型。支持分区建树的软件包括:RaxML,MrBayes和Beast等。
作者选择了以下数据集进行 PartitionFinder 分析(见图 S2):
- 首先,对超级矩阵 A(基因数据块)和超级矩阵 A(蛋白结构域数据块)进行了分隔方案分析和比较,以便根据校正后的 Akaike 信息准则确定哪一个更适合进一步分析。超级矩阵 A(基因数据块)由 1,478 个基因数据块组成,这些数据块是将转录组数据分配到 1:1 的直系同源群中得到的;而超级矩阵 A(蛋白结构域数据块)由 2,868 个数据块组成,这些数据块包括注释的蛋白质结构域族、结构域和基因空白区域。(为了在 PartitionFinder 中进行分析,作者将超级矩阵 A(结构域数据块)中长度小于 21 个氨基酸位点的数据块集中到一个数据块中。这样做的原因是,对于如此短的数据块,确定模型的意义不大。)
- 然后,对超级矩阵 B 进行分隔方案分析,删除信息含量=0 的所有数据块(族clans、结构域domains、空白voids)
- 最后,对超级矩阵 C 进行分隔方案分析,从中剔除分类群覆盖率不足的数据块(见第 3.4 章)。
作者改进了PartitionFinder 软件,使得新版 PartitionFinder 使用改进的启发式方法来确定元分区,并使用 RAxML(目前最快的最大似然树重建程序)。此外,PartitionFinder 现在还可以利用多处理器机器,并行启动多个 RAxML 分析。(这部分可以参考这篇文章,比较详细)PartitionFinder2筛选系统发育分析分隔模型.PDF (book118.com)

在这篇文章的分隔方案分析中,分为两个步骤:
- 第一次运行产生分隔方案:
将 PartitionFinder 的搜索范围限制在两个氨基酸替代模型上,即 LG+ GAMMA 和 LG + GAMMA + F(使用基于经验的频率,using empirical based frequencies)。
第一次运行 PartitionFinder 时,在 PartitionFinder 配置文件中使用了以下参数:
branchlengths = linked;
models = LG+G, LG+G+F;
model_selection = AICc;
search = greediest;
命令行参数:
--raxml --cmdline-extras "-T 2" -p 2 --greediest-schemes 100 \
--cluster-weights "1,1,0,1" --greediest-percent 1
- 第二次运行产生最优氨基酸替代模型(经验模型):
最佳模型是从 RAxML中的所有可用模型中确定的。(作者共测试了来自 11 个基础模型的 44 个模型。对于每个基础模型都考虑了 +G、+G+I、+G+F 和 +G+I+F 等子模型。)
在分区查找器配置文件中使用了以下参数:
branchlengths = linked;
models = all_protein;
model_selection = AICc;
search = user;
命令行参数:
--raxml --cmdline-extras "-T 2" -p 4
进化模型(机械模型&经验模型)
进化模型的主要区别在于每次对所考虑的数据集估计的参数数量和对大型数据集一次估计的参数数量。
机械模型将所有排列描述为针对所有分析的数据集估计的大量参数的函数。最好使用最大似然法。这样做的优点是可以根据特定的数据集特征(例如不同的 DNA 成分偏差)定制模型。使用太多参数可能会导致问题,特别是如果参数可以相互补偿(这可能会导致歧义)。在这种情况下,数据集通常太小,无法提供足够的信息来准确估计所有参数。
经验模型是通过估计大型数据集中的多个参数(通常是速率矩阵和字母频率中的所有条目,请参阅上面的 GTR 模型)来创建的。这些参数是固定的并可重复用于所有数据集。这样做的优点是可以更准确地估计这些参数。通常不可能仅从当前数据集估计置换矩阵的所有条目。缺点是从训练数据估计的参数太笼统,可能不太适合您的特定数据集。此问题的一个潜在解决方案是使用最大似然(或其他方法)来估计数据中的某些参数。
替代模型 Substitution Model: 最新的百科全书、新闻、评论和研究 (academic-accelerator.com)
3.7 核苷酸水平的特定位点速率建模
计算特定位点的一致性指数需要树的拓扑结构,多个树形结构有利于减少随机误差,可以使用自举重采样法(boostrap-resampling)生成树库:将串联比对分成多个 150,000 bp 的片段。然后使用 RAxML 为每个片段生成 100 个快速自展ML树。
比对不能完全被 150,000 整除:本文中包括所有密码子位置的超级矩阵 C 的比对最后一段为 190,377 bp,而只包括第 2 个密码子位置的超级矩阵 C 的比对最后一段为 113,459 bp。
把所有自举树都汇集到一个树文件中,并使用 PAUP 中的 "重新加权字符reweight characters "选项计算每个自举树的特定位点一致性指数(site-specific consistency indices)。从自举树库中提取每个位点的最佳(最大)一致性指数。利用上述数值使用 Python 中的scikit-learn 软件包 k-means 聚类算法将位点聚类为离散的分区。
k-means的核心目标是将给定的数据集划分成k个簇cluster/组group(K是超参),并给出每个样本数据对应的中心点。
该思维导图引用自——
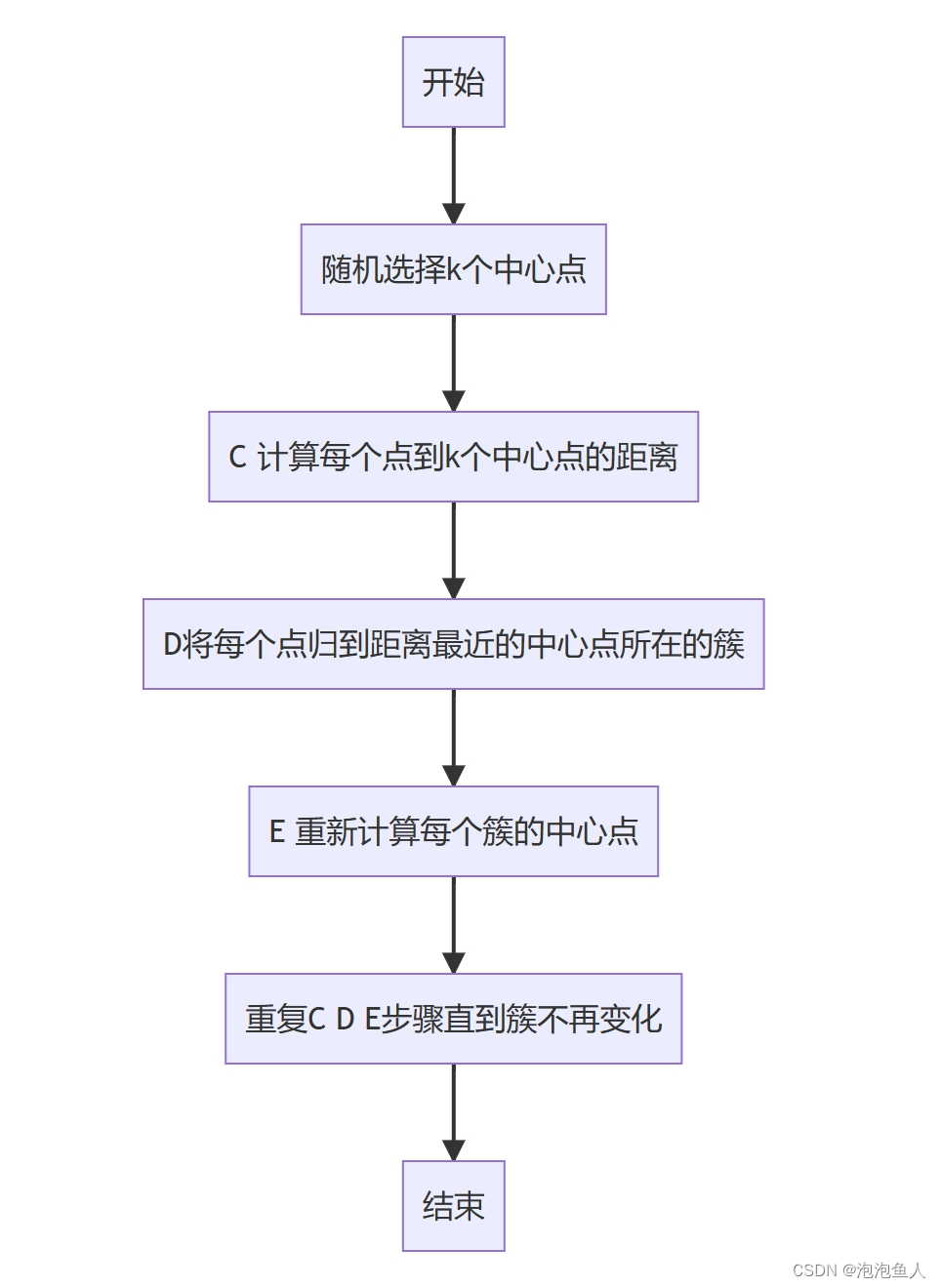
使用 k-means++ 选项生成中心点并运行该算法直至收敛。(选择初始中心点)
该文章中指定了簇的数量:对核苷酸水平上的超级矩阵 C 使用了 50 个聚类(只包含第 2 个密码子位置);对整个超级矩阵 C 的核苷酸排列使用了 100 个聚类(包括所有密码子位置)。
3.8 基于序列的系统发育分析
本文用于推断系统发育关系的方法:ExaML
3.8.1 树搜索
尽管逐步加和步骤是随机的,但随机逐步加和的解析树往往产生相同的起始树。因此作者使用了完全随机的起始树和随机逐步加和解析树,以避免任何系统性偏差。
根据 PSR 模型和 GAMMA 模型得到的树,都要根据 GAMMA 模型进行评分,当其中一方的整体对数似然得分明显更高时,使用得分更高的模型,当并没有明显的整体对数似然得分更高的模型时,将这两种模型都纳入分析。
作者对每个超级矩阵都总共进行了 50 次 ML 树搜索,在搜索最佳树时采用了不同的策略:
- 使用随机逐步加和解析起始树进行 5 次 ML 树搜索,使用完全随机的起始树进行 20 次 ML 树搜索,并采用 PSR(每个位点速率) CAT 模型。然后,根据 GAMMA 变异速率模型,使用四个离散速率类别discrete rate categories和速率的中位数对得到的 ML 树进行评分。
-
使用随机逐步加和解析起始树进行 5 次 ML 树搜索,以及基于完全随机的起始树进行 20 次 ML 树搜索,并使用中位数和四个离散速率类别的 GAMMA 变异速率模型。
ML树是所有 50 个树推论中总体对数似然函数值得分(log-likelihood score)最高的树。
PartitionFinder 为每个分区选择的最优氨基酸替代模型见补充文本第 2.6.1 章(超级矩阵 B:770 个分区,比对长度为 576 840 个氨基酸位点;超级矩阵 C:479 个分区,比对长度为 413 459 个氨基酸位点;超级矩阵 D:311 个分区,比对长度为 201 000 个氨基酸位点)
其中只选择了超级矩阵 C 来根据核苷酸数据重建系统发育树。一个只使用第二位密码子,另一个考虑了所有编码位置。这两个核苷酸数据集都是根据特定位点速率建模方法的结果进行分区的,而不是使用用 PartitionFinder 优化的基于蛋白质结构域的分区方案。(含有第二个密码子位置的核苷酸数据集包括 50 个分区,而含有所有密码子位置的核苷酸数据集包括 100 个分区)
用于 ML 树推断的详细命令如下(其中所有的起始树都是由RAxML生成的)
#生成随机逐步加法解析起始树:
(1) $ raxmlHPC-SSE3 -X -s supermatrix.phy -n starting_tree -p \
$RANDOMSEED -m MODEL#生成完全随机的起始树:
(2) $ raxmlHPC-SSE3 -d -y -s supermatrix.phy -n starting_tree\
-p $RANDOMSEED -m MODEL氨基酸数据集的模型 (-m) 设置为 PROTGAMMALG,核苷酸数据集的模型 (-m) 设置为 GTRGAMMA。
所有 ML 树搜索(每个数据集 50 次)均使用 ExaML 进行。在 ML 树搜索过程中,对每个元分区分别独立优化了速率异质性模型的 alpha 形参数、核苷酸替换 GTR 模型的速率以及用于氨基酸数据的 PSR 模型和用于核苷酸数据的 GAMMA 模型的每个位点速率类别。对所有分区的分支长度进行了联合优化。(如何优化?)
使用 ExaML 软件包中提供的解析器,根据超比对F(superalignments)和分区信息边界(partition information boundaries)生成了相应的二进制(计算机可读)文件,作为在 ExaML 中进行系统发育分析的输入。随后分别使用 PSR 或 GAMMA 比率异质性模型进行 ML 树搜索:
#使用速率异质性的PSR模型(25个类别)和相应的起始树进行ML树搜索:
(3)$ examl-AVX -m PSR -s supermatrix.binary -t \
RaxML_starting_tree -n output_PSR -Q#使用速率异质性的 GAMMA 模型进行ML树搜索
(4) $ examl-AVX -m GAMMA -s supermatrix.binary -t \
RAxML_starting_tree -n output_GAMMA -Q -a使用 ExaML 和速率异质性 GAMMA 模型离散近似值的中值(选项 -f E -a)对得到的 50 个树形拓扑的参数和分支长度(如上所述)进行了优化。
#参数和分支长度的优化给出了最佳拓扑:
(5) $ examl-AVX -f E -m GAMMA -n output -s supermatrix.binary \
–t ML_tree -Q -a
3.8.2 自举法检验(Bootstrap Replicates)
依据RAxML-Light手册的指引在ExaML中计算非参数自举重复
(参见https://github.com/stamatak/RAxML-Light-1.0.5)
关于自举重复的次数,作者采用了 MRE 自举收敛标准取代随机重复,这种方法也被称为 "自举停止" (bootstopping):在 50 次自举搜索后,使用加权标准(加权罗宾逊-福尔德Robinson Fould距离)评估收敛性,并使用标准 RAxML 构建多数规则扩展(MRE)一致树(autoMRE, 使用默认阈值 0.03)。继续运行 50 次自举直到收敛。根据这种 "自举停止 "标准,选择支持度稳定性与自举重复次数和计算资源相关的最有效方法。
#确定自举重复是否已收敛(满足自举停止条件):
(6) $ raxmlHPC-SSE3 -n output_autoMRE -z batch_bootstrap_trees\
–I autoMRE -m MODEL#在最佳 ML 树上绘制自举bootstraps support支持度:
(7) $ raxmlHPC-SSE3 -n output_support -z batch_bootstrap_trees\
-f b -m MODEL -t best_tree使用 Treegraph v. 2 ,选择蜱 Ixodes scapularis 作为外群,对所有树进行置根,并进行美化以便发表。
3.9 Rogue Taxon 分析
Rogue taxon的定义是在一组树(例如自举树)中占据了多个位置的类群(或被表述为位置不确定的类群),在推断进化关系的过程中带来不确定性,使我们不能准确的下定科学结论。其存在会导致原本稳定的进化关系中出现低自举支持度或不被支持的结果。
作者开发并使用开源工具 RogueNaRok识别自举树集合中的rogue taxa。RogueNaRok用于计算删除rogue taxon后一致树(consensus tree)支持率的提高幅度。其结果取决于 drop set 参数,该参数指定了 RogueNaRok 算法在每一步中考虑修剪的最大类群数量。
t75 setting可提高至少 75% 支持率的枝的数量;每一个数据集都用逐步增大的dropset(1-10)进行十次搜索,只采纳支持度总体提高最大的结果。
从自举树中剪除已被确定为 "rogue"的分类群(见补充文本,第 2.8 章,表 S24),并在剪除的最佳 ML 树上绘制 BS 支持度图。作者没有修剪弱的rogue taxon。如果rogue taxon的总体改进值低于 0.3,将其归类为 "弱 "类群。改进值(improvement value)的定义是原始一致树与已剪除rogue taxon的一致树之间的累积自举支持率之差。
















 3155
3155











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









