







上节我们完成了Space Ranger分析,在输出结果binned_outputs文件夹内默认生成了2um,8um,16um bin的结果,这节我们使用10x文章中推荐的8x8um bin结果进行后续分析。为了演示,我们取P1_CRC样本数据进行后续分析(多样本合并一般都需要进行去批次处理,如果有需要,后续我们会继续分享多样本合并分析步骤)。
主要步骤:
1.读取8um bin结果;
2.数据质控;
3.细胞过滤;
4.降维聚类;
5.特征基因筛选
环境加载
import os
import numpy as np
import pandas as pd
import scanpy as sc
import seaborn as sns
import omicverse as ov
import matplotlib.pyplot as plt
from matplotlib.pyplot import rc_context
import warnings
warnings.filterwarnings('ignore')
plt.style.use('default')
plt.rcParams['figure.facecolor'] = 'white'
sc.settings.set_figure_params(dpi=100, dpi_save=200, figsize=(5, 5), facecolor='white')
1. 8um bin数据读取
数据读取使用scanpy的read_visium函数即可,要注意一点,Visium HD使用的Space Ranger v3.0为了节省输出文件空间,将之前的spatial文件夹下的tissue_positions_list.csv文件格式变成了tissue_positions.parquet,为了使用read_visium函数正确读取数据,首先要将tissue_positions.parquet文件转成tissue_positions_list.csv文件。
data_dir = './CRC/P1_CRC/'
tissue_position_df = pd.read_parquet(f'{data_dir}/binned_outputs/square_008um/spatial/tissue_positions.parquet')
tissue_position_df.to_csv(f'{data_dir}/binned_outputs/square_008um/spatial/tissue_positions_list.csv', index=False, header=None)
adata = sc.read_visium(f'{data_dir}/binned_outputs/square_008um', library_id='P1')
adata.obs['sample'] = 'P1'
adata.var_names_make_unique()2. 数据质控
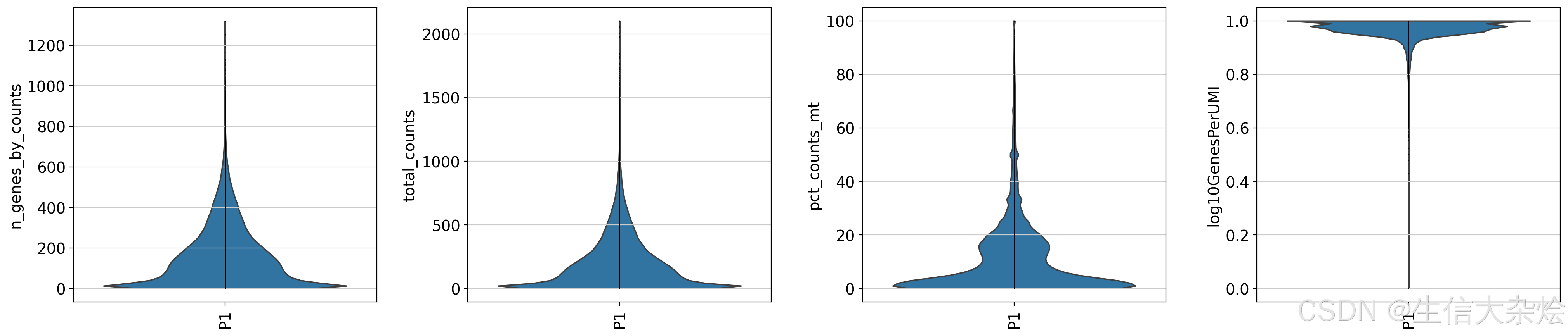
空间转录组学实验中不可避免会引入一些技术噪音,比如由于实验处理或数据测序中的技术误差,导致部分细胞的测序数据异常(例如某些细胞可能由于实验处理不当或实际转录活性低,表现为转录本数量非常少)。细胞QC的目的是识别和过滤掉这些异常细胞,以避免它们对下游分析(如差异表达分析、聚类或功能分析)产生负面影响。通过去除低质量的细胞,我们可以确保保留下来的细胞是转录活性正常、具有代表性的细胞。这样不仅可以提高分析结果的生物学意义,还能减少假阳性结果。
# Data QC
adata.var['mt'] = adata.var_names.str.upper().str.startswith('MT-')
adata.var['ribo'] = adata.var_names.str.upper().str.startswith(("RPS","RPL"))
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt', 'ribo'], percent_top=None, log1p=False, inplace=True)
adata.obs['log10GenesPerUMI'] = np.log10(adata.obs['n_genes_by_counts']) / np.log10(adata.obs['total_counts'])
sc.pl.violin(
adata,
['n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'log10GenesPerUMI'],
groupby='sample',
jitter=False,
rotation=90,
multi_panel=True,
show=False
)
3. 细胞过滤
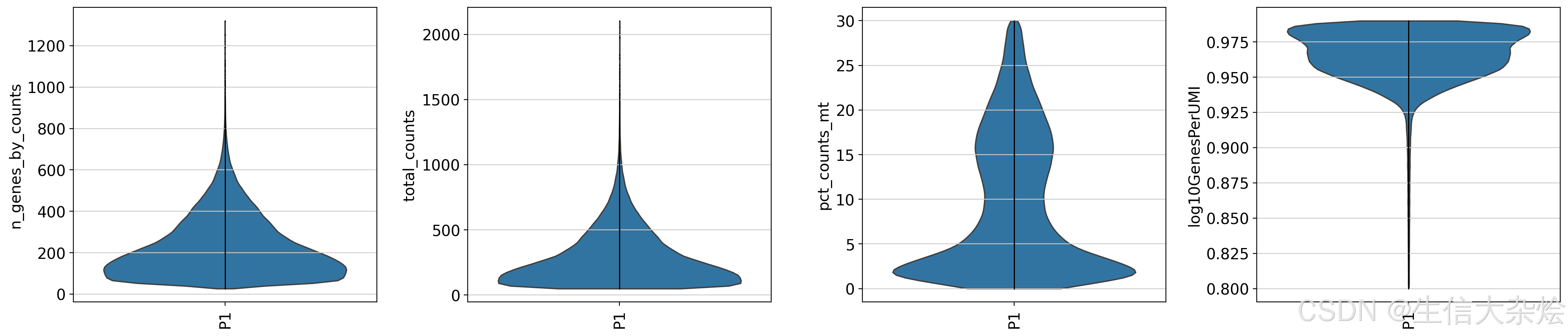
低质量细胞会影响后续分析结果,这里我们和单细胞数据分析一样,首先将低质量细胞进行去除(当然也有些人不进行过滤,直接后续分析),这里我们将8um bin内鉴定到UMI counts数小于50个的细胞、线粒体基因占比大于30%的细胞、以及log10GenesPerUMI(主要反映的是细胞中鉴定到基因文库的复杂性)值小于0.8的细胞认为是低质量细胞,进行去除。
## Cell filter
sc.pp.filter_cells(adata, min_counts=50)
adata = adata[(adata.obs['pct_counts_mt'] < 30) & (adata.obs['log10GenesPerUMI'] > 0.8) & (adata.obs['log10GenesPerUMI'] < 0.99)]
sc.pl.violin(
adata,
['n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'log10GenesPerUMI'],
groupby='sample',
jitter=False,
rotation=90,
multi_panel=True,
show=False
)
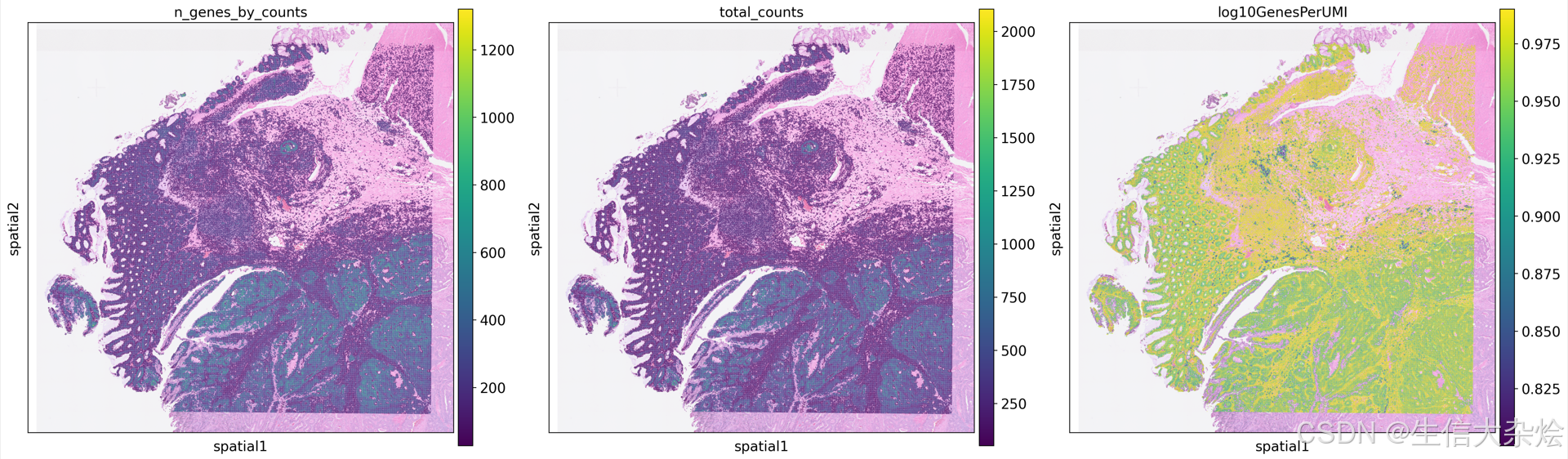
with rc_context({'figure.figsize': (8, 8)}):
sc.pl.spatial(
adata,
color=['n_genes_by_counts', 'total_counts', 'log10GenesPerUMI'],
library_id='P1',
alpha_img=0.6
)
细胞过滤后,空间上展示展示基因数、counts数等
4. 降维聚类
经过过滤之后的表达矩阵,使用Scanpy 进行降维聚类分析:
1)首先进行数据的表达量均一化,对测序深度进行校正,消除技术噪音;
2)选取高可变基因(细胞间高特征基因),用于提高细胞分群准确度(默认值为2000);
3)进一步对表达量值均一化处理后,利用PCA分析对数据进行降维,PCA降维是一种线性降维方法,运用方差分解,将高维的数据映射到低维的空间中;然后基于SNN聚类算法对细胞进行聚类和分群,构建细胞间的聚类关系;
4)最后将降维后的数据传递到UMAP进行可视化展示,细胞之间的基因表达模式越相似,在UMAP图中的距离也越接近。
## data normalization and log10 transform
adata.layers["counts"] = adata.X.copy()
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
adata.raw = adata
## select highly variable gene and data scale
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5, n_top_genes=2000)
sc.pl.highly_variable_genes(adata, show=False)
adata = adata[:, adata.var['highly_variable']]
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
## PCA
sc.tl.pca(adata, svd_solver='arpack')
sc.pl.pca(adata, color='sample', show=False)
sc.pl.pca_variance_ratio(adata, log=True, show=False)
## UMAP
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=15)
sc.tl.umap(adata)
sc.tl.leiden(adata, resolution=0.6, key_added='res_0.6')
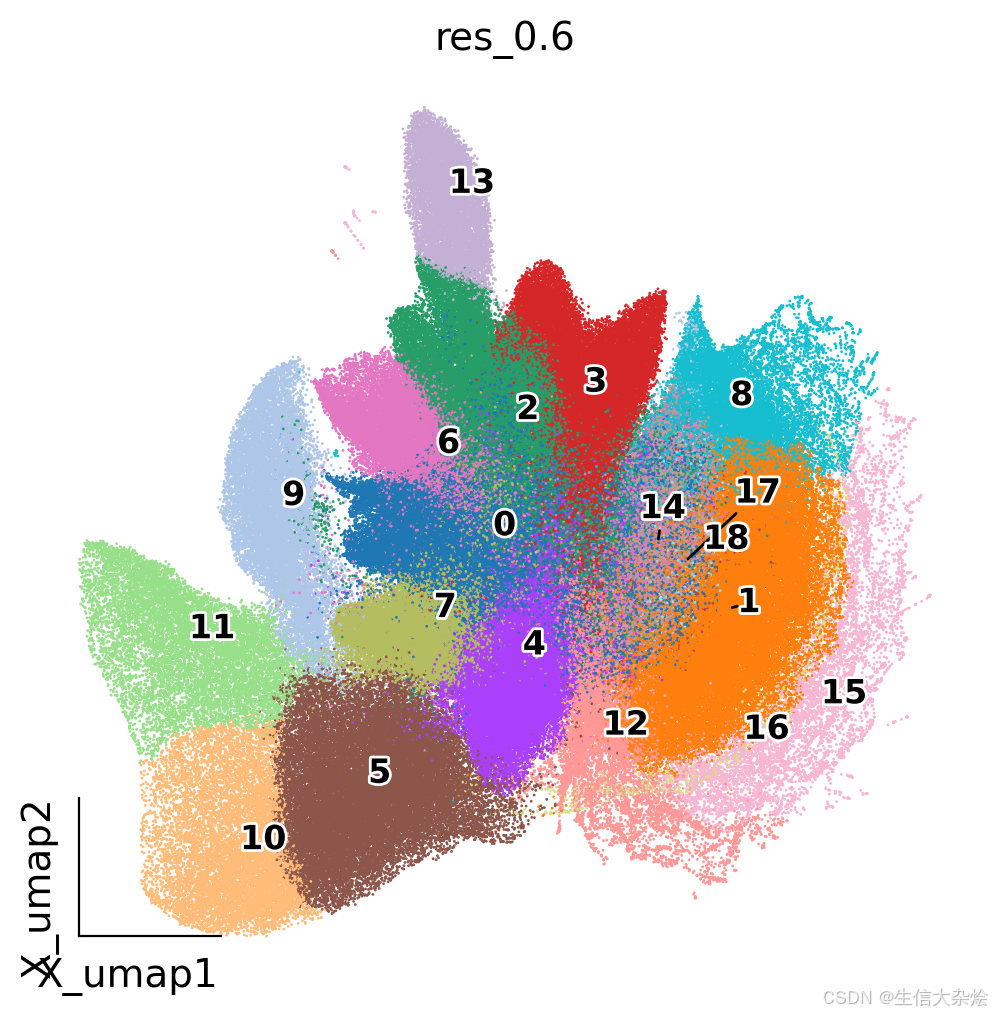
sc.tl.leiden(adata, resolution=0.8, key_added='res_0.8')降维聚类结果UMAP及空间展示
from matplotlib import patheffects
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(6,6))
ov.pl.embedding(adata,
basis='X_umap',
color=['res_0.6'],
size=4,
show=False,
legend_loc=None,
add_outline=False,
frameon='small',
legend_fontoutline=2,
ax=ax
)
ov.utils.gen_mpl_labels(
adata,
'res_0.6',
exclude=("None",),
basis='X_umap',
ax=ax,
adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')),
text_kwargs=dict(fontsize= 12 ,weight='bold', path_effects=[patheffects.withStroke(linewidth=2, foreground='w')] ),
)
单细胞降维聚类结果展示
降维聚类结果空间展示
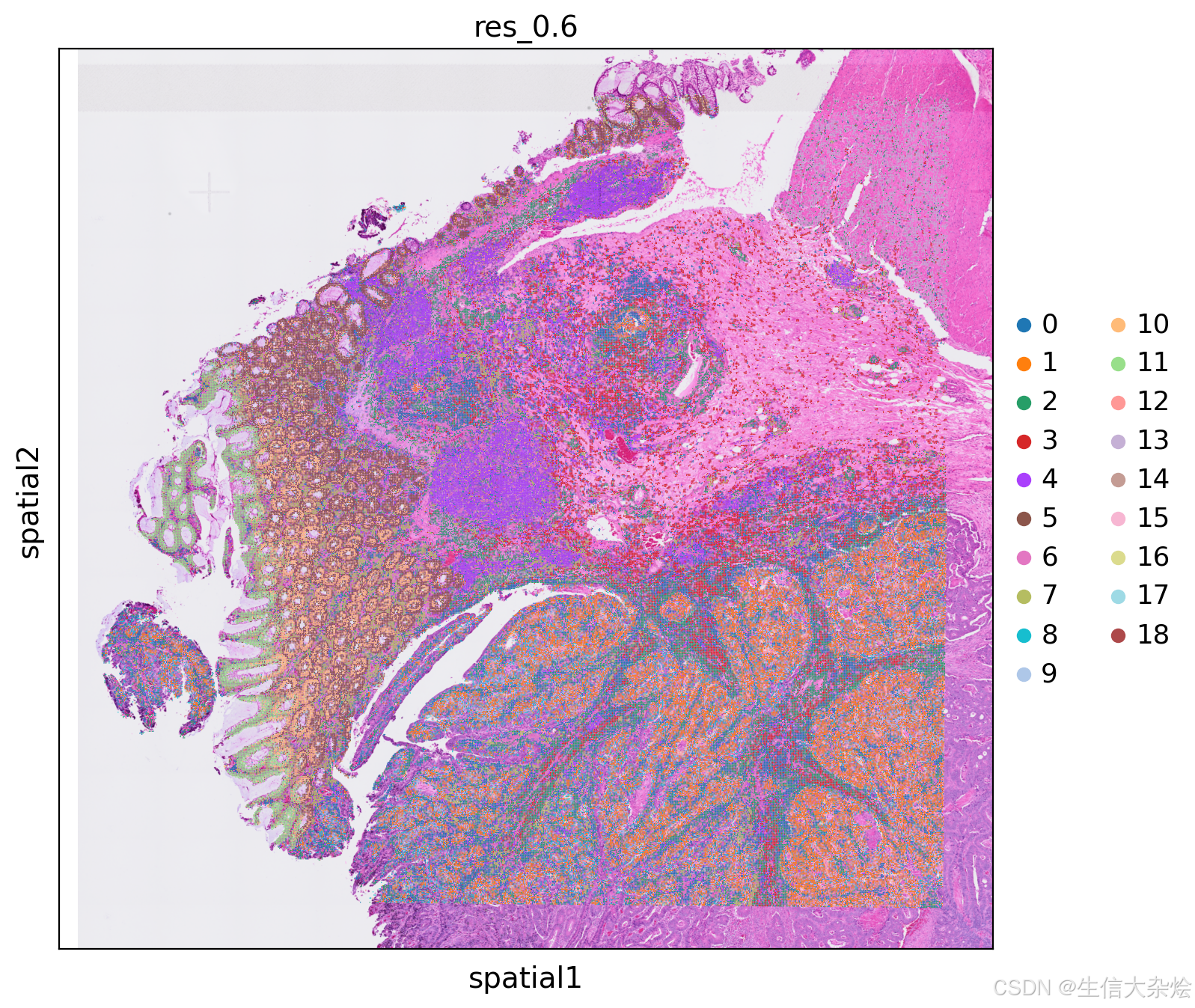
特征基因筛选
sc.tl.rank_genes_groups(
adata,
groupby='res_0.6',
use_raw=True,
method="wilcoxon",
pts=True
)
sc.pl.rank_genes_groups_dotplot(
adata,
groupby='res_0.6',
standard_scale="var",
n_genes=5,
cmap='bwr',
show=False,
)
result = adata.uns['rank_genes_groups']
groups = result['names'].dtype.names
all_diff = []
for c in groups:
tmp = sc.get.rank_genes_groups_df(adata, group=c)
tmp = tmp[(tmp['logfoldchanges']>0) & (tmp['pvals_adj']<0.05)]
tmp['cluster'] = c
all_diff.append(tmp)
all_diff = pd.concat(all_diff, axis=0)各细胞Cluster Marker基因Dotplot, 结合上一步各Cluster特征基因,进行细胞类型定义
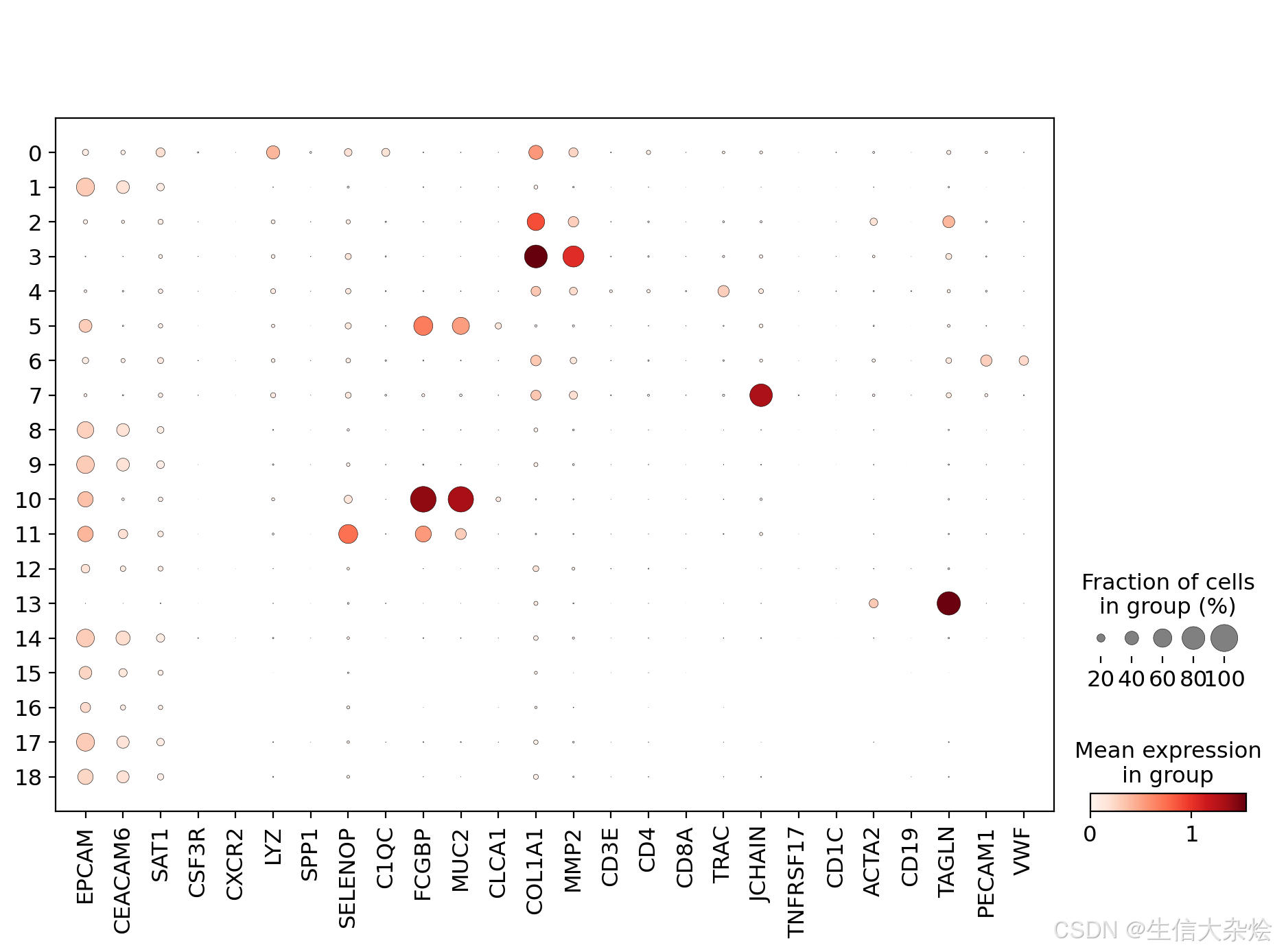
微信公众号:生信大杂烩

















 530
530

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









