







本文来自“生信算法”公众号。
序列比对(sequence alignment)又称序列联配, 为确定两个或多个序列之间的相似性(similarity) 或同源性(homology) ,将序列按照一定规律进行排列的操作。序列相似性和序列同源性为两个不同的概念,序列相似性是可以量化的参数,是一个数量值,如相似性为90%。而同源性指两条序列是否来自共同祖先,来自同一祖先则为同源序列。一般而言,序列相似性越高,其同源可能性越大。
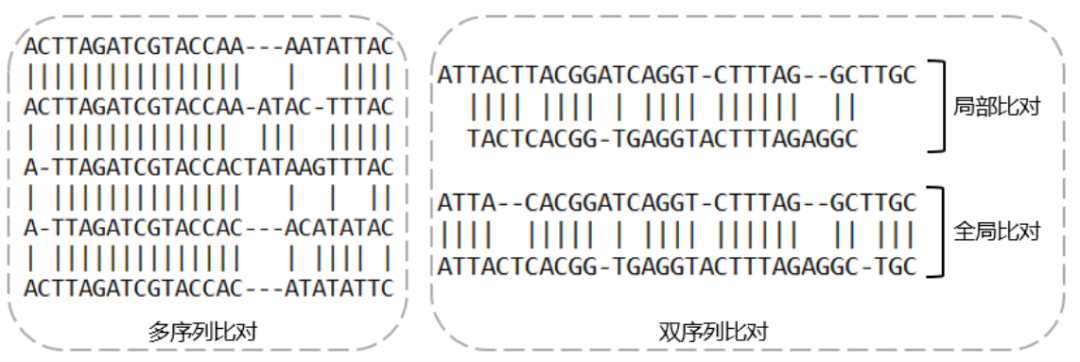
序列比对经过多年的发展, 已提出了一系列算法,其中Needleman-Wunsch算法(NW算法)是全局比对的经典算法,而Smith-Waterman算法(SW算法)是局部比对的代表性算法。NW算法和SW算法都采用动态规划算法实现序列联配。二代、 三代序列比对算法基本上利用NW和SW算法得到序列比对结果。
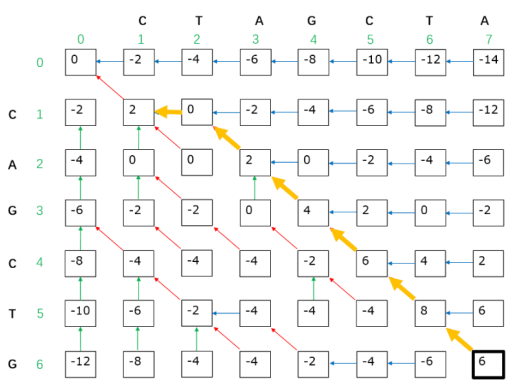
序列比对是宏基因组数据分析的基础,但非常耗时,尤其是对于长序列。随着测序技术的发展,尤其是第三代单分子测序技术的出现,产生的序列长度平均值可以超过20000 bp。如此长的序列比对,需要消耗更多的计算时间和计算内存。为了提高长序列比对速度,Martin Sosi等人2017年在Bioinformatics期刊上发表了一篇快速序列比对软件包Edlib,可实现长序列,甚至长基因组之间的序列比对。

如何使用 Edlib 序列比对包
Edlib主要基于Myers’s bit-vector (位相量)实现序列比对过程,而且采用了带状比对(banded alignment)思想,可以进一步提高运算速度和降低内存消耗。Edlib实现过程比较复杂,不容易理解,对计算机数据结构、编程技巧有较高的要求。本次不做详细解读,主要高速大家怎么用 Edlib 软件包。Edlib是用C++语言编的,主要包含两个文件:edlib.h 头文件和edlib.cpp源文件。
首先在Github上下载Edlib源代码:
git clone https://github.com/Martinsos/edlib.git
然后用CMake命令进行安装:
cd build && cmake -D CMAKE_BUILD_TYPE=Release .. && make然后在 bin/ 目录中会生成二进制文件:
执行以下命令进行测试:
.runTestsEdlib运行时候需要包括"edlib.h"文件,如下:
#include <cstdio>
#include "edlib.h"
int main() {
EdlibAlignResult result = edlibAlign("hello", 5, "world!", 6, edlibDefaultAlignConfig());
if (result.status == EDLIB_STATUS_OK) {
printf("edit_distance('hello', 'world!') = %d\n", result.editDistance);
}
edlibFreeAlignResult(result);
}Edlib主程序实现函数为edlibAlign(上面第5行),需要5个参数,其中第一个是比对的第一条序列,第二个参数为序列的长度,第三个是第二条序列,第四个是序列的长度,第五个是比对模式设置,一般用默认值。
Edlib比对模式(EDLIB_MODE)有三种设置,分别为NW、SHW和HW模式。比如我们有两条序列需要进行比对:
s1= GCTAACTGGC 和 s2=CGTAGCTG
NW模式就是我们之前的全序列比对,SHW模式是从序列 s1 开始,只比对到序列 s2的末端,HW是寻找序列 s2 和序列 s1 之间最相似的部分,也就是半全局(semi-global sequencealignment )比对。
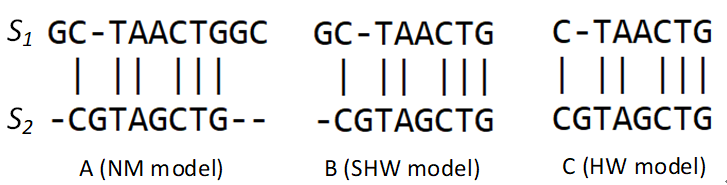
具体实现不同的比对模式如下图第5个参数:EDLIB_MODE_HW
edlibAlign(seq1, seq1Length, seq2, seq2Length, edlibNewAlignConfig(42, EDLIB_MODE_HW, EDLIB_TASK_PATH, NULL, 0));
比对结果
Edlib和其他2个方法SeqAn和Parasail进行了比较,从下表中可以看出,比对的序列长度都是10000 bp起步,Edlib实现的序列比对速度明显高于其他方法,尤其是序列相似性越高,比对的速度越快。

参考资料:
- Šošić M, Šikić M. Edlib: a C/C++ library for fast, exact sequence alignment using edit distance. Bioinformatics, 2017, 33(9): 1394-1395.















 338
338











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









