







本文来自“生信算法”公众号。

2021年12月3-5号,有幸参加第六届中国计算机学会生物信息学会议并做分会场报告,会议地点在山东青岛。报告题目是:smsMap: mapping single molecule sequencing reads by locating the alignment starting positions。是我们去年的做的一个小工作,一个简单的三代序列比对算法。可以帮助入门者学习,有需要发小文章或者着急毕业的同学可以进行相应改进并发表文章。

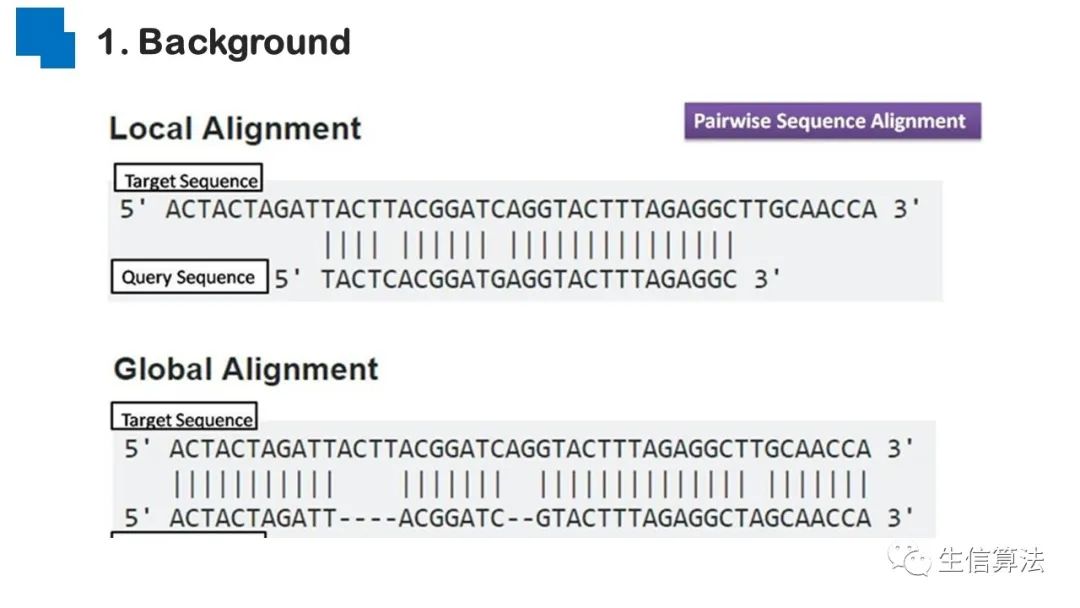
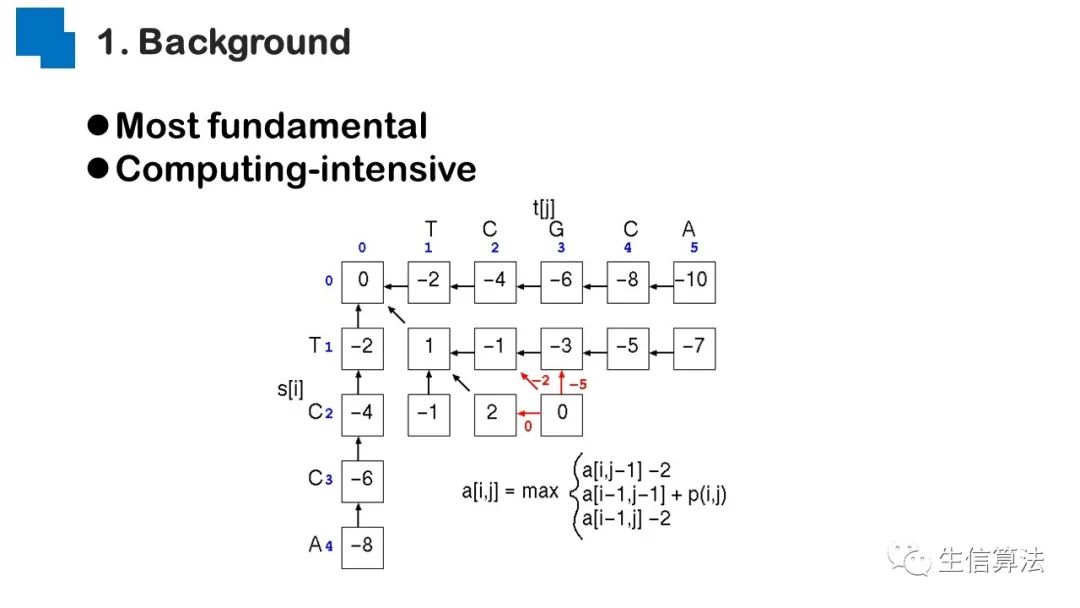
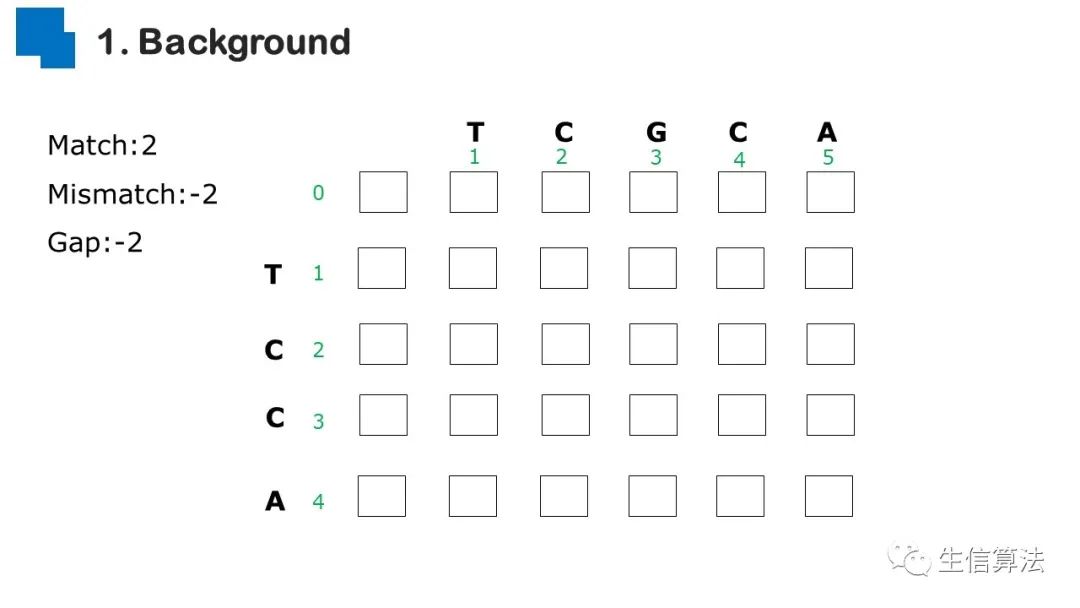
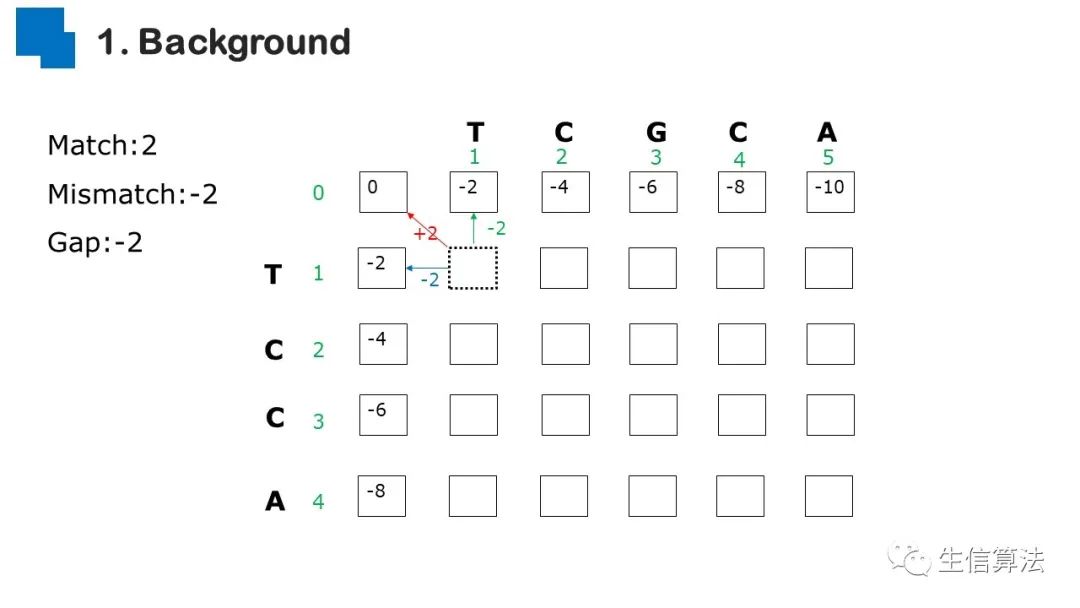
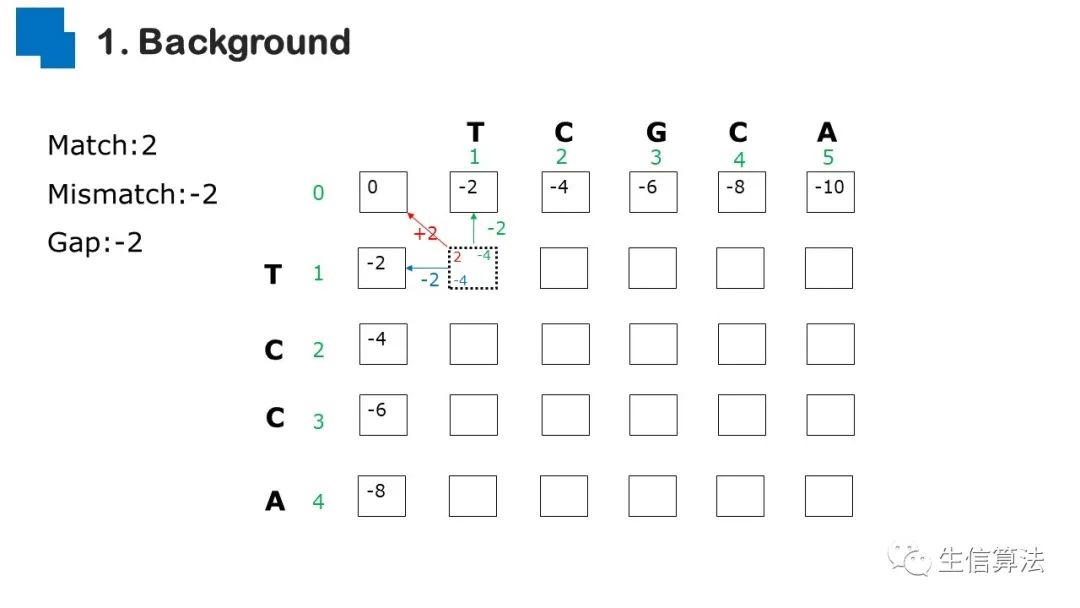
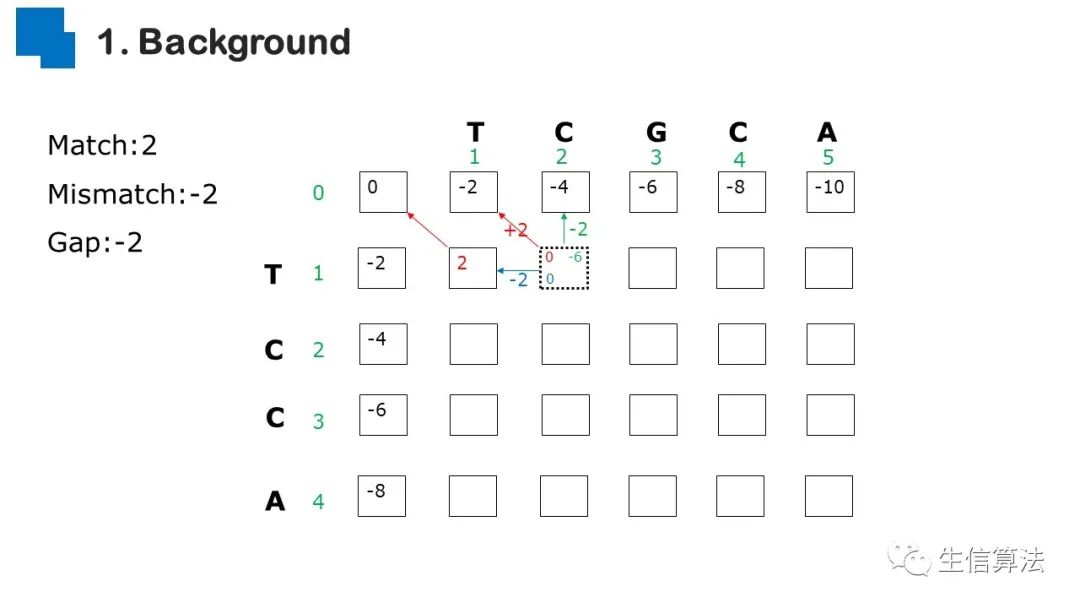
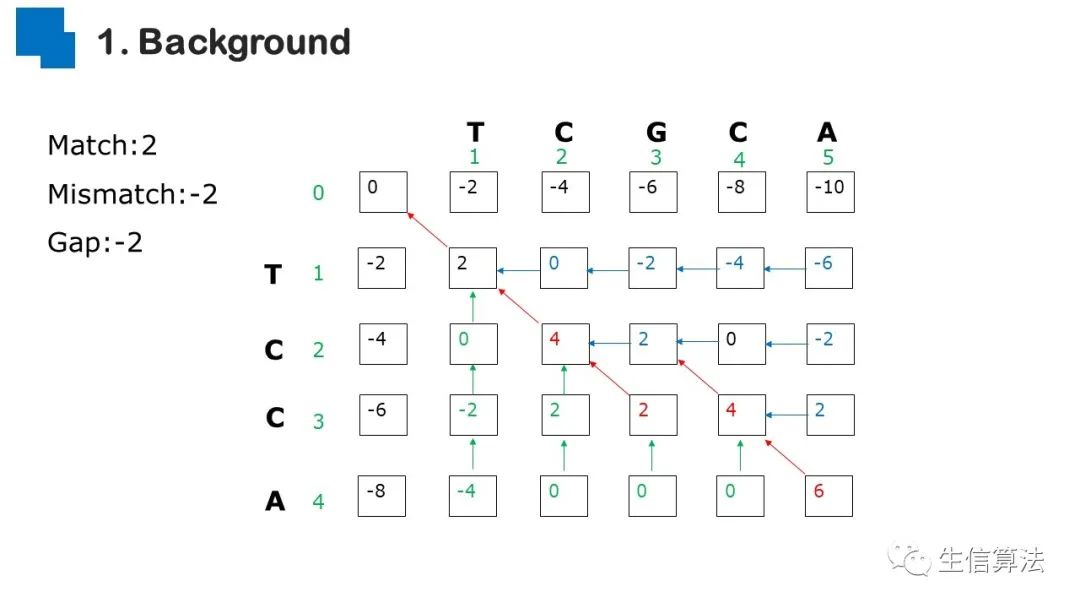
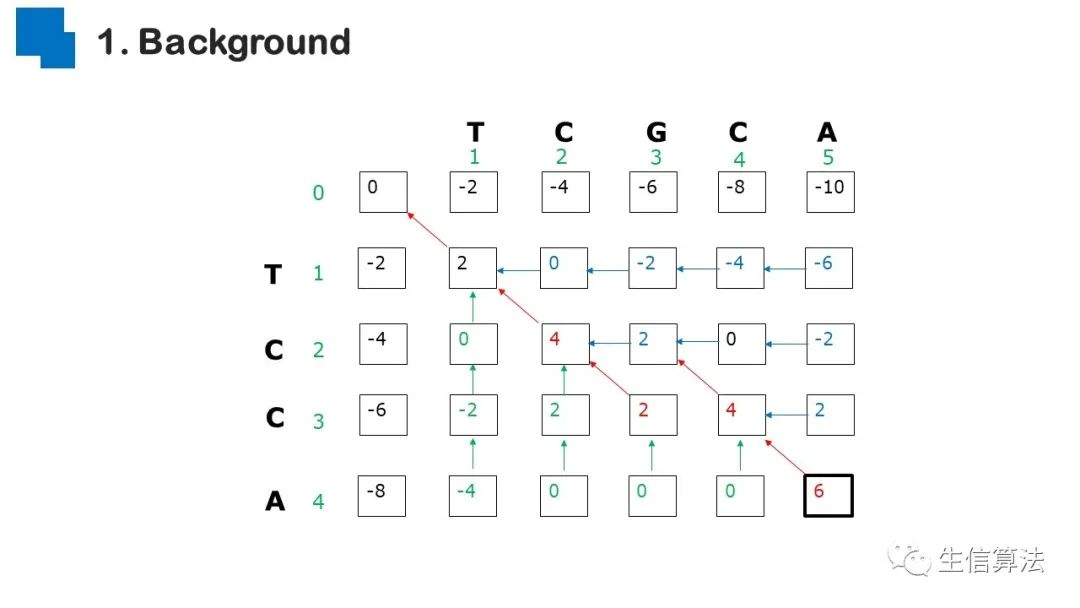
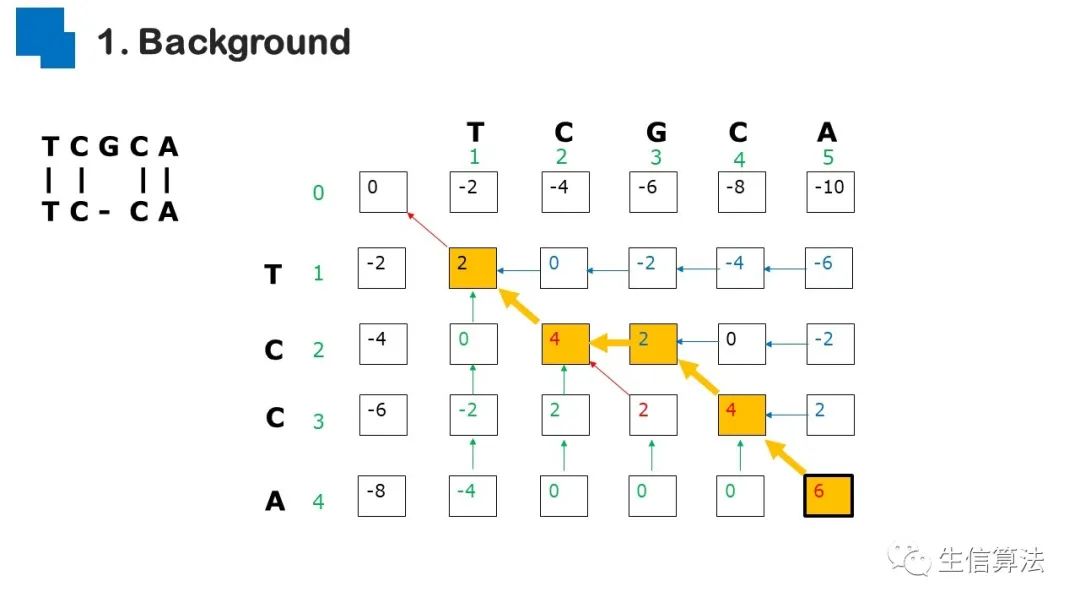
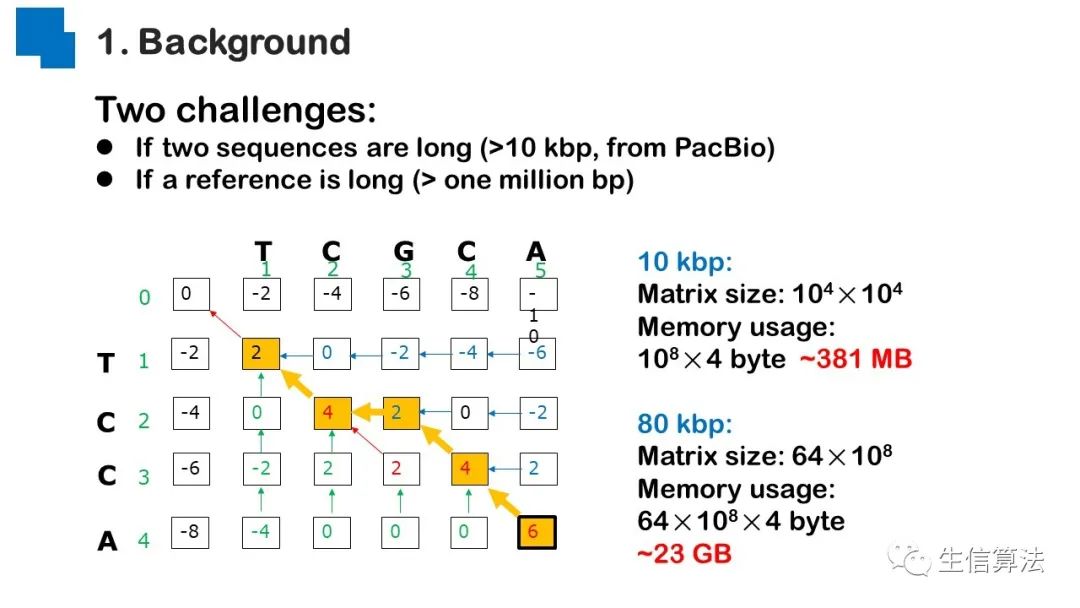
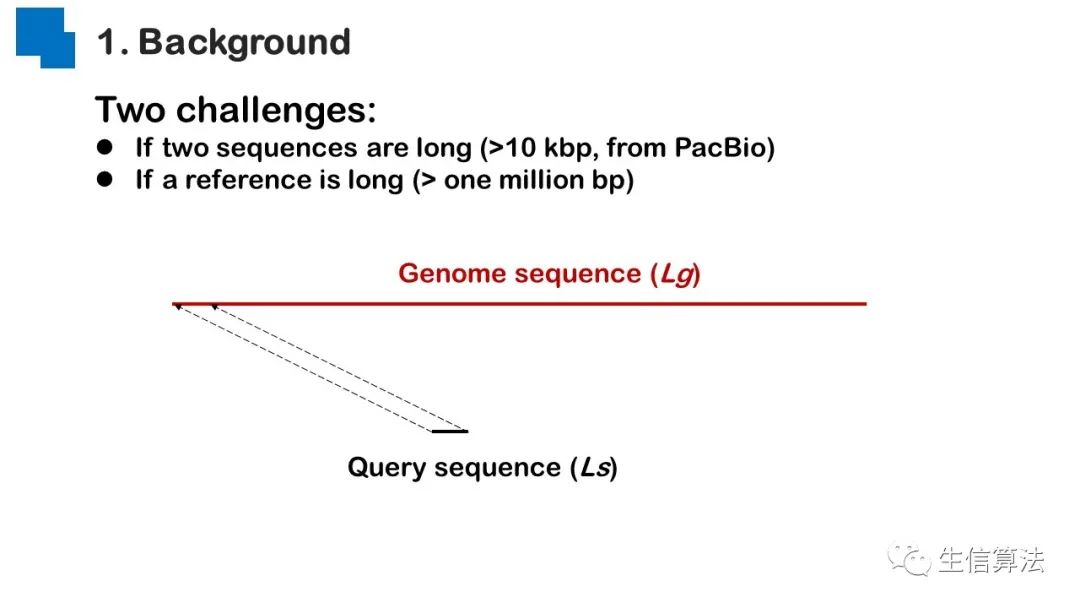
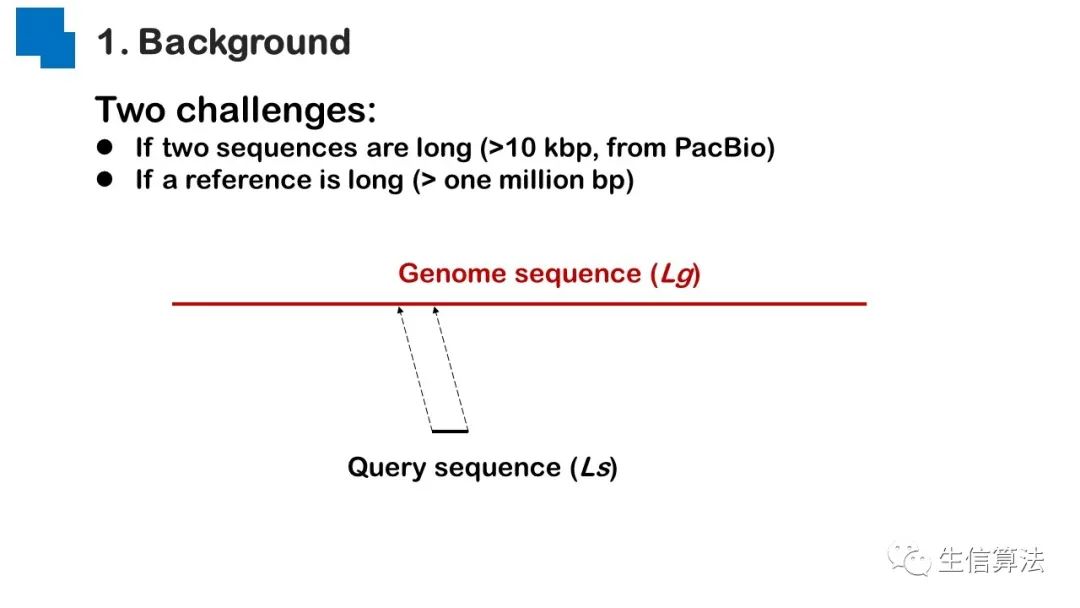
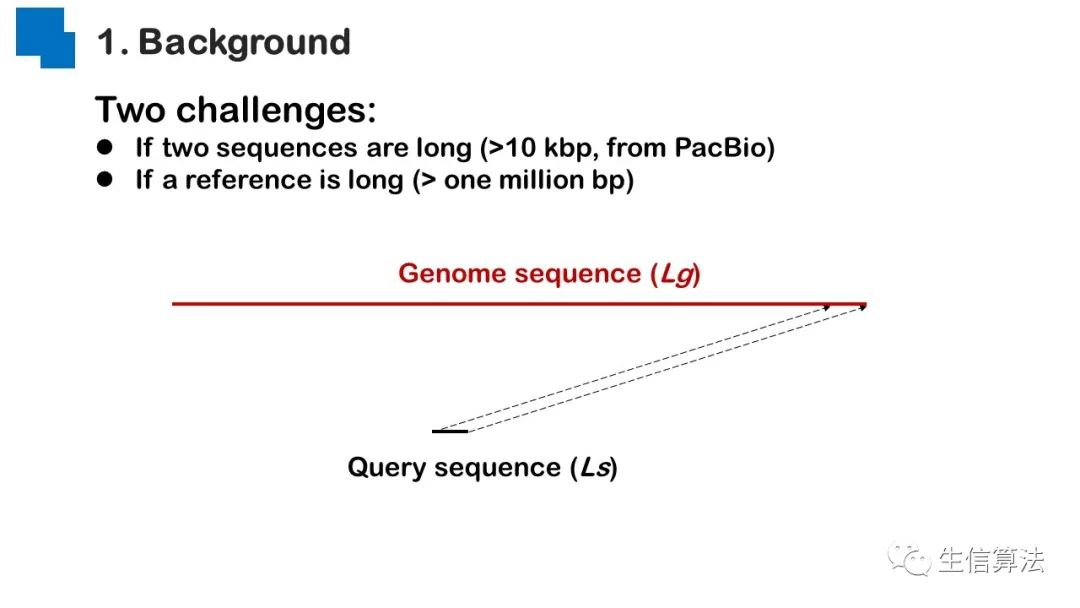
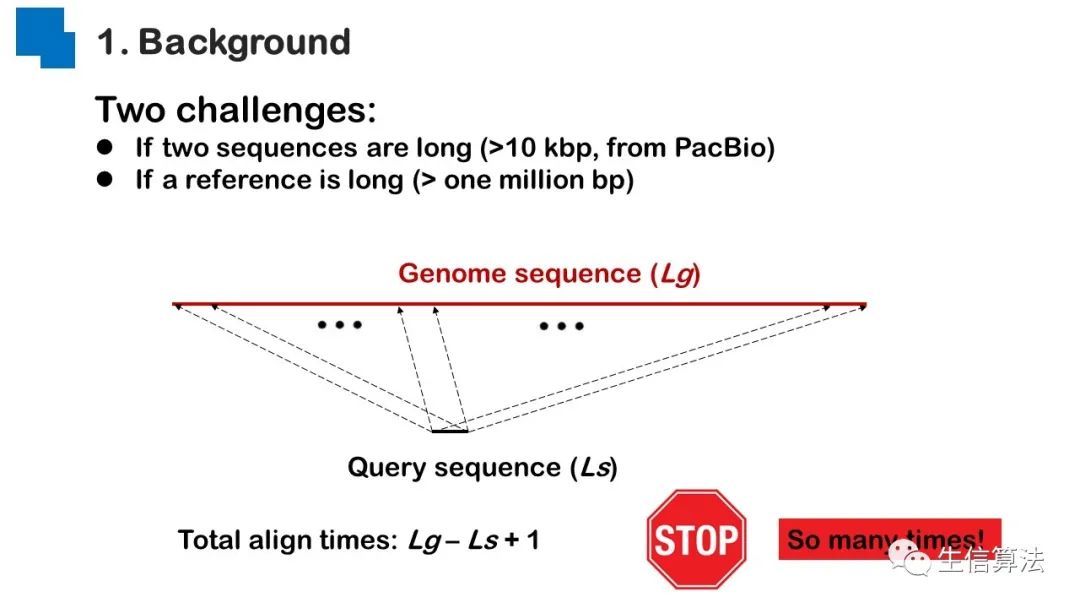
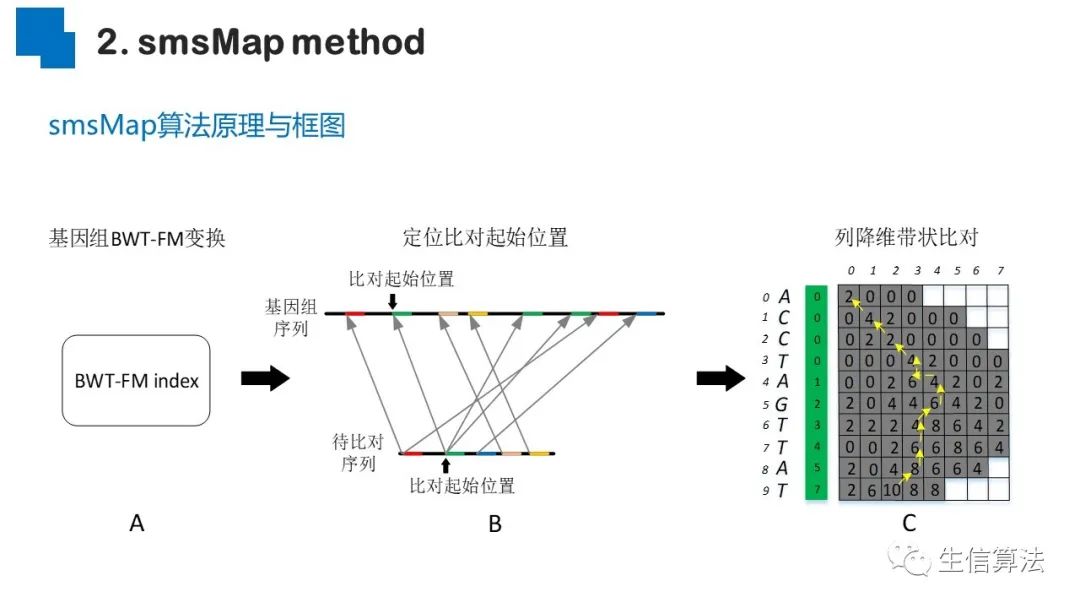
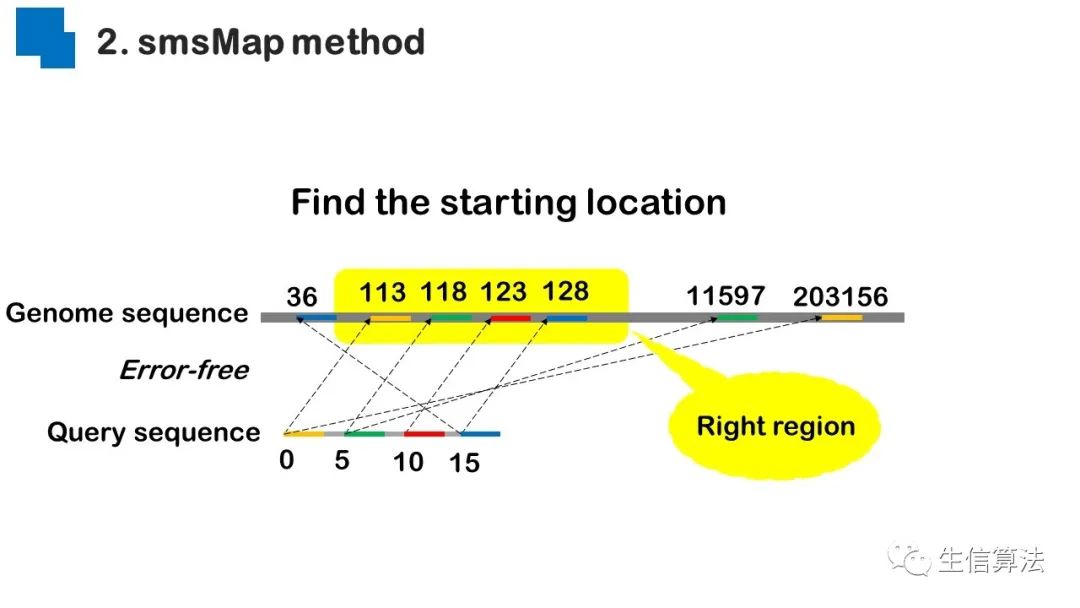
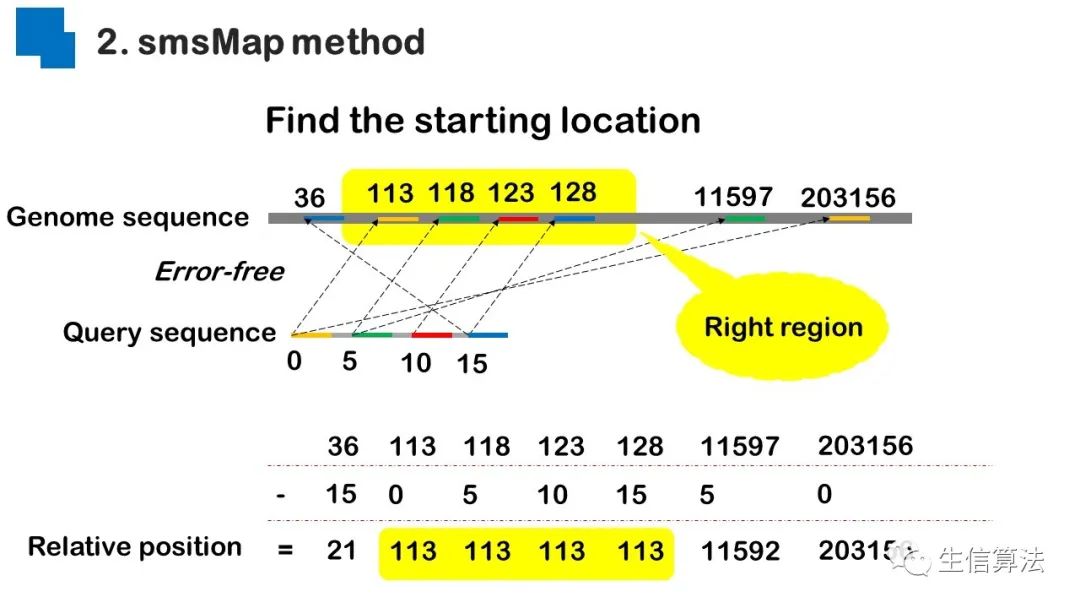
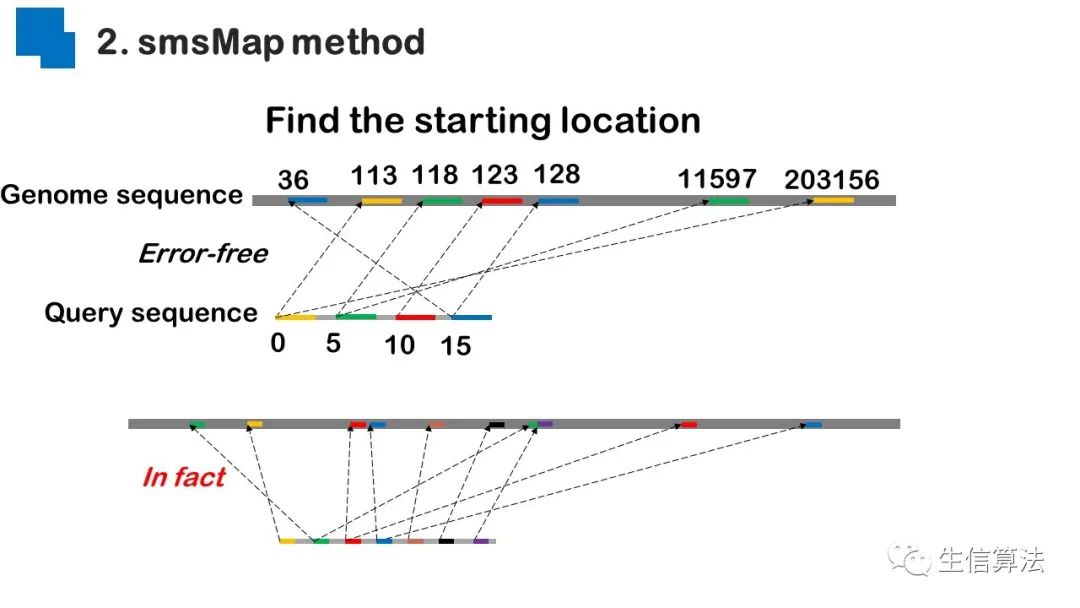
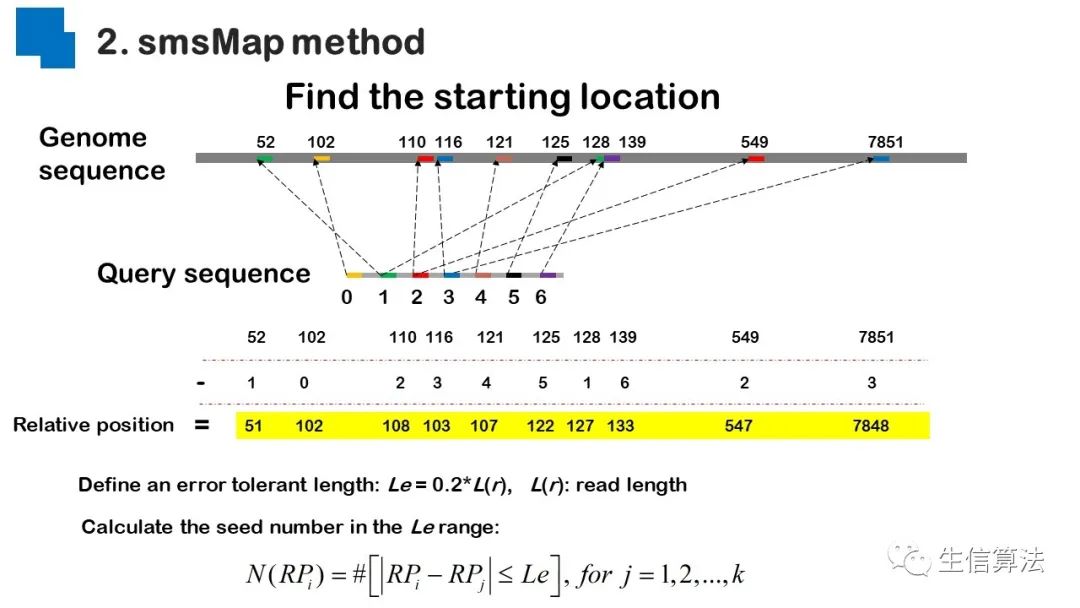
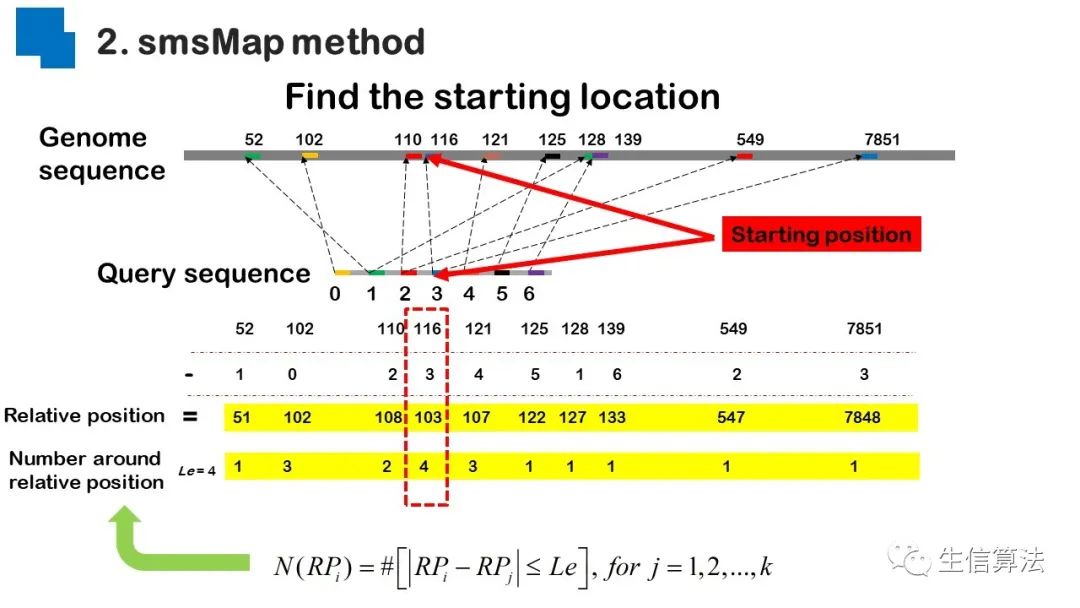
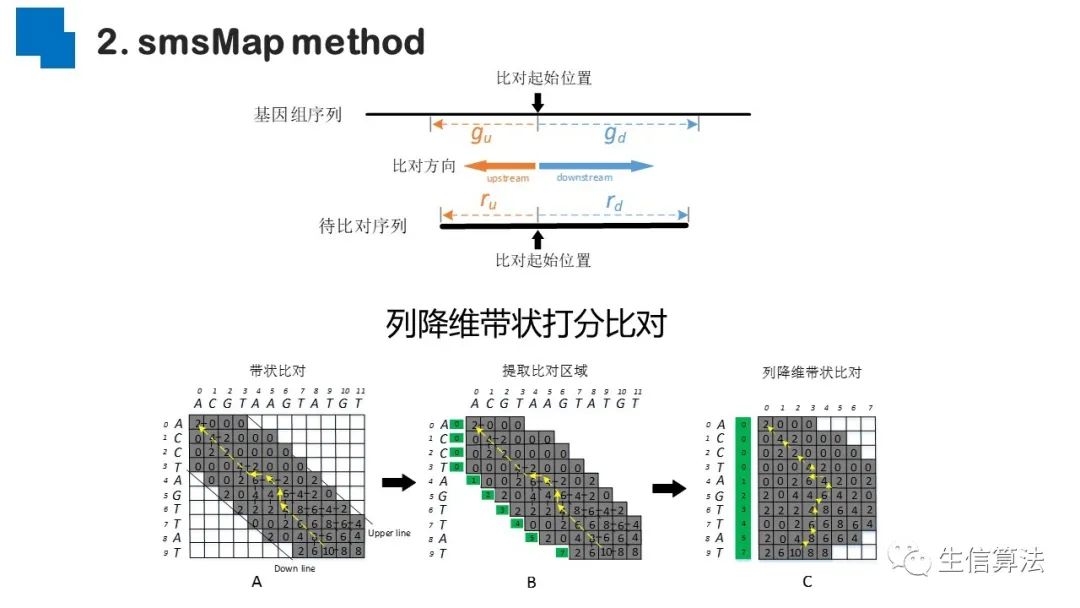
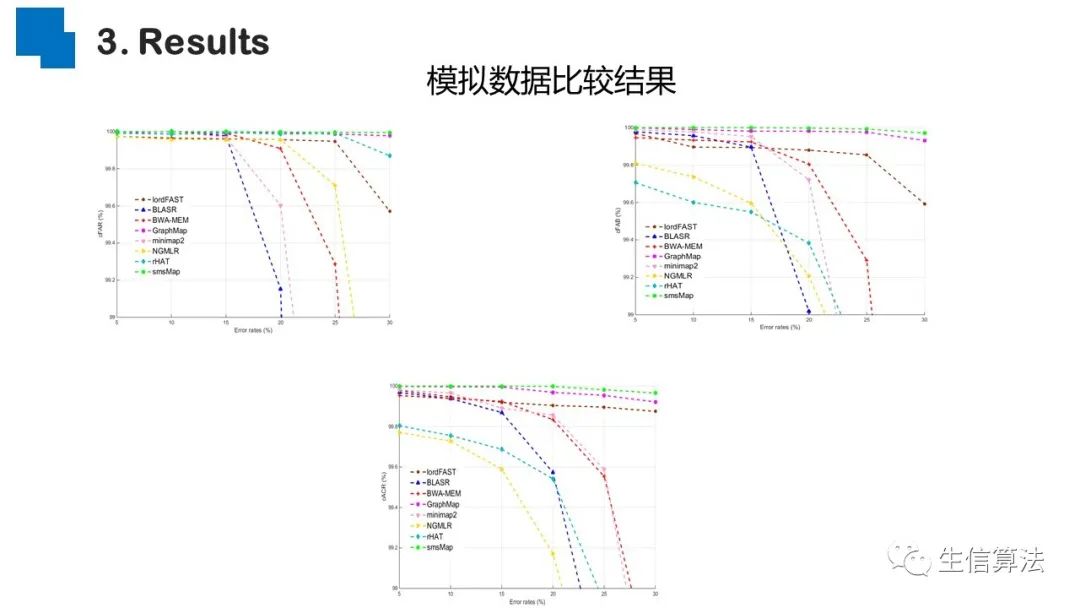
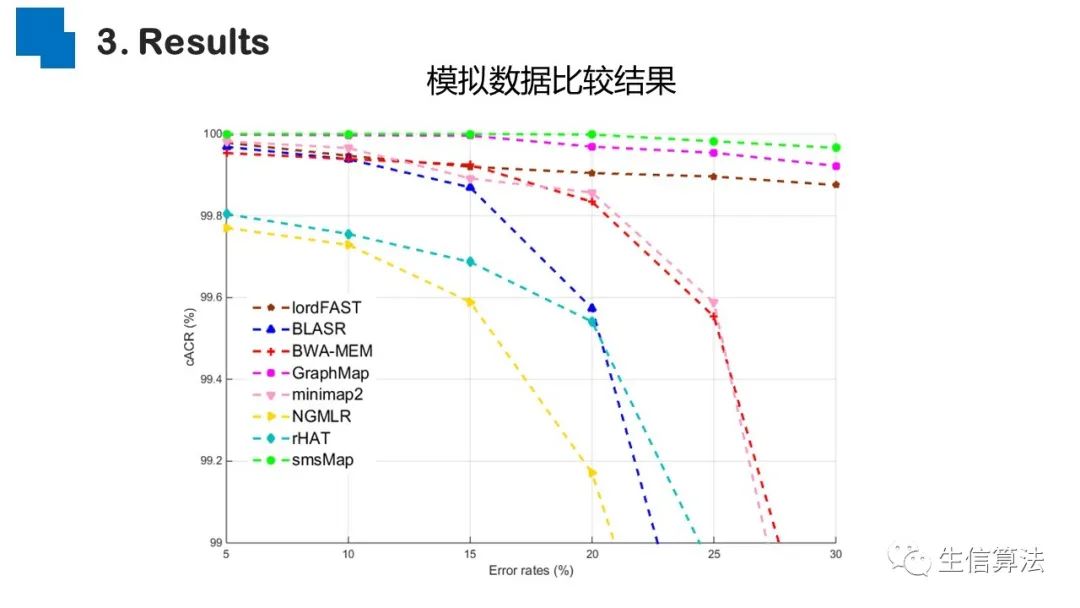
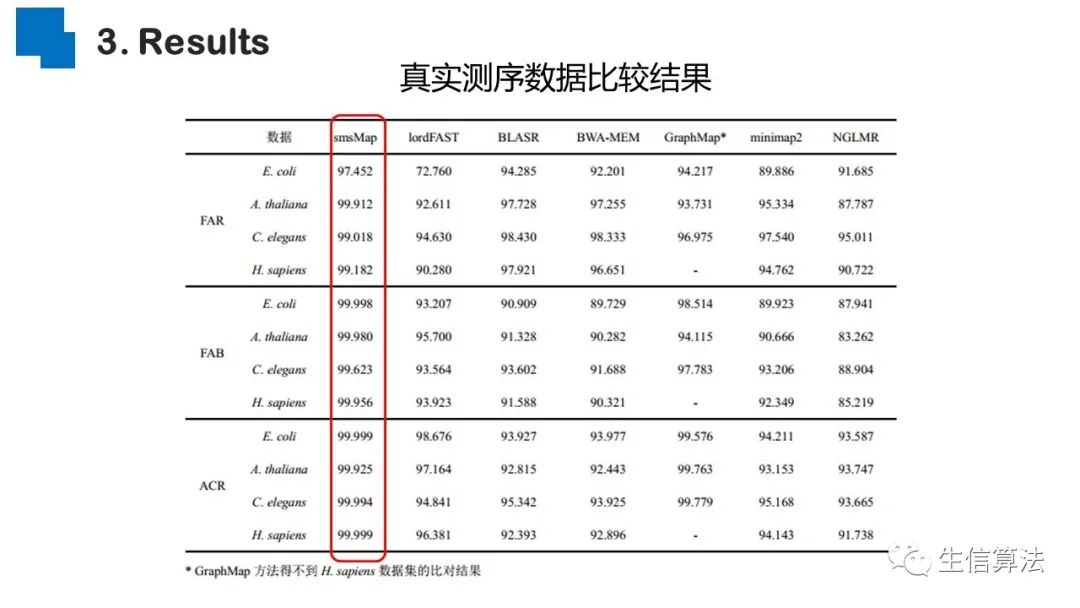
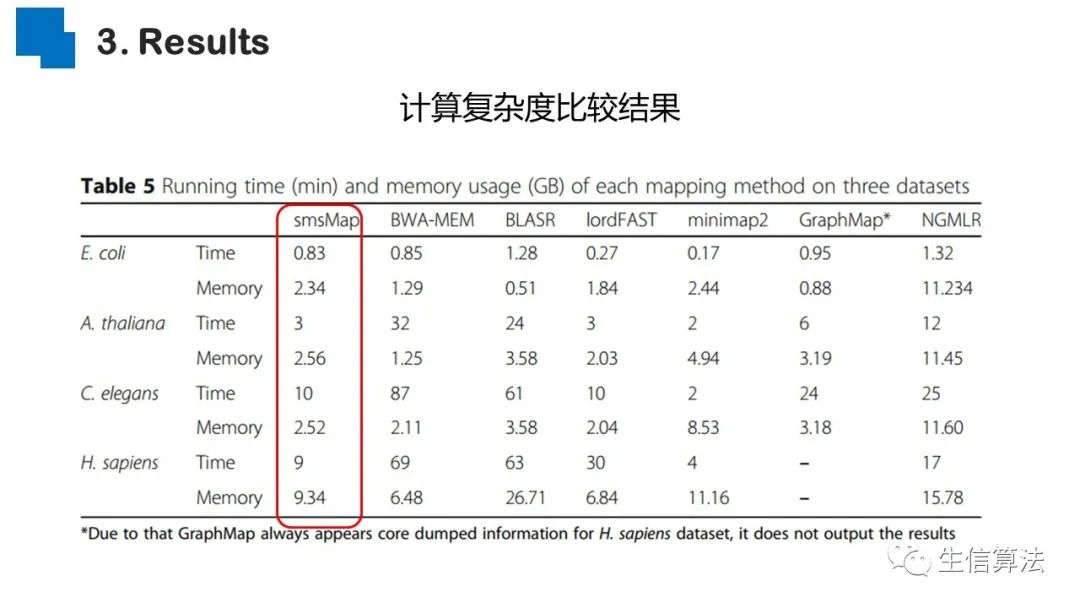
序列比对是生物数据最基本的操作,但同时也是最耗时的,因为需要构建打分矩阵。三代测序序列长度长,错误率高,需要针对性的开发三代序列比对算法。目前三代序列主要采用“种子-延伸”思想,但其大多数序列比对算法只得到局部高质量比对结果,没有得到“end-to-end”的比对结果,比对覆盖率较低,为此,我们开发了一款基于种子定位的三代序列比对算法smsMap,可以提高比对覆盖率,同时对测序错误有一定的鲁棒性。
以下是我的报告PPT,希望对同学问有所帮助,有什么问题可以在后面加我微信互相交流。也恳请各位专家老师批评指正。












总结
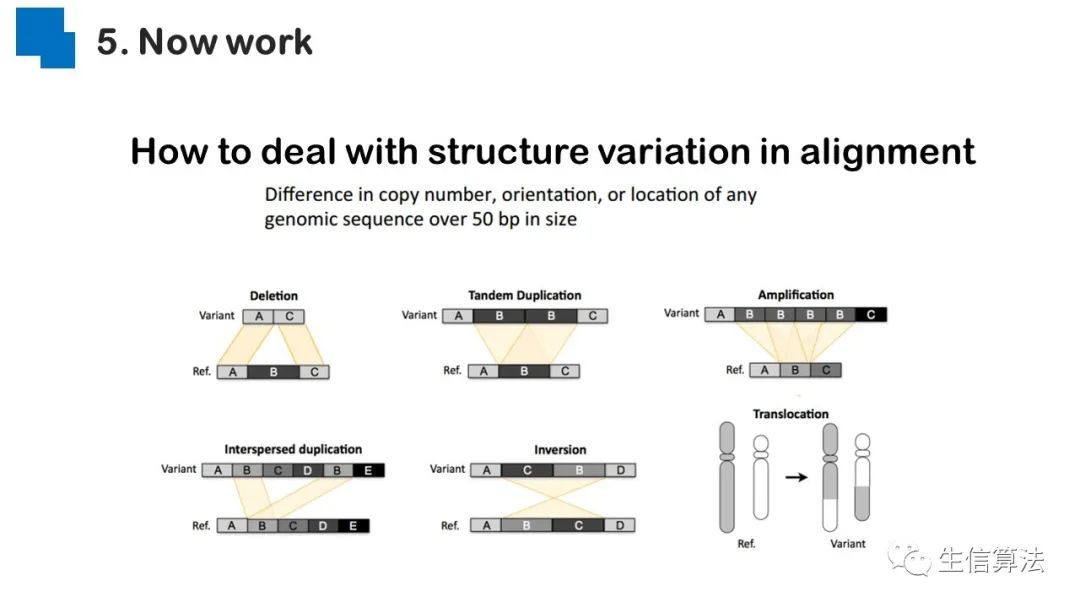
目前我们还在沿着这个工作在做,主要是如何处理结构变异体的问题,以及如何有效检测结构变异体。这篇文章生物信息的内容很少,纯粹是以一个计算机角度的思路在做,还是缺乏生物方面的知识。希望看到本篇文章的生物背景的同学或者老师可以相互交流,你有问题,我们有解决方法,期待合作

。
参考资料:
- Wei Z G , Zhang S W , Liu F . smsMap: mapping single molecule sequencing reads by locating the alignment starting positions. BMC Bioinformatics, 2020, 21(1).



















 7542
7542

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









