







✨【元素魔方学术俱乐部】✨
👩🏫👨🏫我们创建了一个学术交流群
给全国各地以及各种研究方向的硕博
和老师们提供一个交流的平台📚🧪
感兴趣的话欢迎加入
📲本公众号中回复“社群”
会自动发送二维码,扫码就能加入啦
群内可以交流学术问题🔬,或者交交朋友
要是搞网恋的话我们也不拦你,还会帮你💘💪!
论文简介

标题:Reaction-induced unsaturated Mo oxycarbides afford highly active CO2 conversion catalysts
作者:Xingtao Sun1,2, Jiafeng Yu 1 , Habib Zada1,2, Yu Han1,2, Ling Zhang1,2, Huaican Chen3,4, Wen Yin2,3,4 & Jian Sun 1
期刊:nature chemistry(IF:19.2)
原文链接:
https://www.nature.com/articles/s41557-024-01628-4
研究背景
由于二氧化碳过量排放引发的气候变化问题日益严重,开发经济且可持续的二氧化碳利用技术已成为科学界的关键任务。逆水煤气变换反应(RWGS)是将二氧化碳转化为一氧化碳的有效途径,所生成的一氧化碳可进一步在费托合成等下游过程中转化为多种高价值的液体燃料和化学品。然而,现有的催化剂系统存在许多挑战,例如贵金属催化剂(如Pt、Ru、Ir等)由于成本高昂不适合大规模应用,过渡金属催化剂(如Fe、Co、Ni等)则容易产生甲烷副产物,增加了分离处理的难度。因此,开发一种具有高活性、高选择性和稳定性的催化剂系统,并且以丰富的地球元素为原料,成为实现大规模CO2转化的迫切需求。
钼基碳化物催化剂由于其独特的原子排列,展示了在不依赖贵金属的情况下实现CO2活化的潜力。然而,传统的钼碳化物制备方法往往需要苛刻的氨化和碳化处理,制备能耗高,同时催化剂的稳定性和结构在反应过程中也存在复杂变化。因此,本研究的目的是开发一种反应诱导的钼氧碳化物催化剂系统,通过反应过程中的原位碳化,去除高能耗的预处理步骤,实现CO2高效转化,并为相关催化体系的工业应用提供新的思路。
论文速览
这篇论文探讨了基于钼氧碳化物的高效二氧化碳转化催化剂的设计及其在逆水煤气变换反应(RWGS)中的应用。在研究背景中,由于二氧化碳排放引发的气候变化问题,开发经济、可持续的二氧化碳转化技术成为重要课题。现有的贵金属和过渡金属催化剂系统要么成本高昂,要么易生成副产物,不适合大规模应用。因此,基于丰富的地球元素的钼碳化物催化剂成为研究重点。
在研究方法上,论文采用了火焰喷雾热解法(FSP)制备Ir掺杂的钼氧化物催化剂,并通过原位表征技术(如X射线吸收谱和中子衍射等)来研究催化剂的结构变化及活性位点。同时,作者利用密度泛函理论(DFT)计算揭示了钼氧碳化物表面的反应机制,尤其是碳循环路径在CO2还原中的作用。
研究结论显示,通过反应诱导形成的钼氧碳化物能够提供高度不饱和的活性位点,促进CO2的高效转化。这种催化剂在600°C的高温下稳定工作超过2000小时,表现出优异的选择性和活性,解决了传统催化剂体系的诸多问题。该研究为钼基催化剂的高温应用及大规模CO2转化提供了新的设计思路。
图文导读
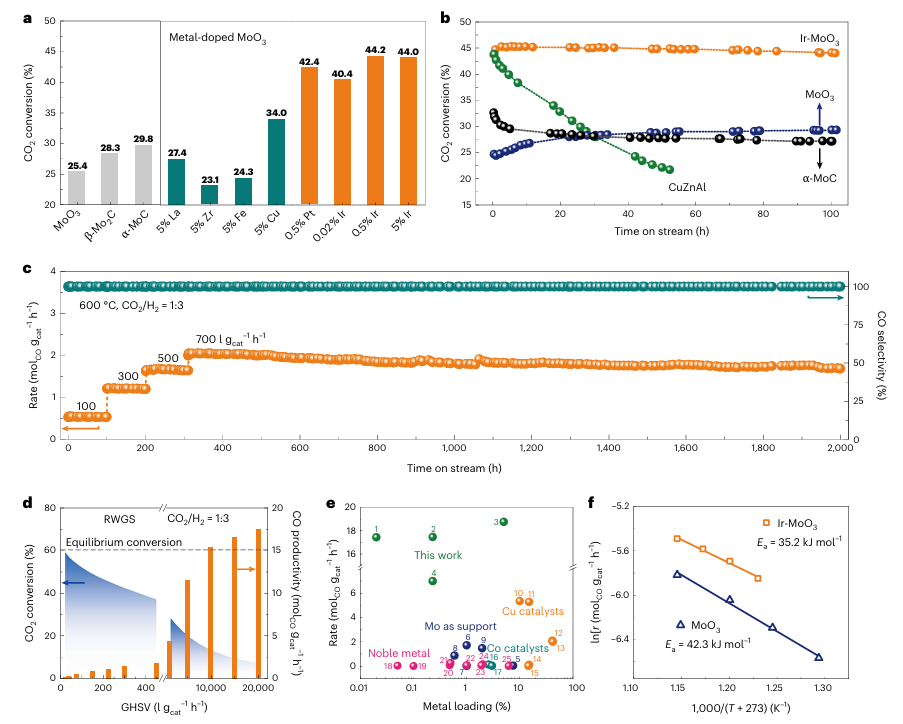
图1 |RWGS反应中的催化性能 a. MoO₃、α-MoC 和 β-Mo₂C 的 CO₂ 转化率,以及通过 FSP(火焰喷雾热解)方法掺杂不同非贵金属(La、Zr、Fe 和 Cu)和贵金属(Pt 和 Ir)的 Mo 基催化剂的性能表现。所有测试的催化剂均在 400°C 下通过 H₂ 预还原 2 小时,并在 600°C 的 RWGS 反应中测试 4 小时。b. 不同催化剂的 100 小时稳定性测试。反应条件为:600°C,0.1 MPa;24% CO₂,72% H₂,4% N₂;气体时空速(GHSV)= 300 l gcat⁻¹ h⁻¹。c. Ir-MoO₃ 催化剂在不同 GHSV 条件下的 2,000 小时长期稳定性测试。d. Ir-MoO₃ 在 600°C 下不同 GHSV 条件下的 CO₂ 转化率和 CO 生产率。e. 本研究中的 Ir-MoO₃ 催化剂性能(绿色)与文献中不同类型催化剂的性能比较,包括 Cu 基催化剂(橙色)、Co 基催化剂(青色)、Mo₂C 和 Mo₂N 支撑的金属催化剂(蓝色)及贵金属催化剂(红色)。具体性能在补充表1中列出。f. 在动态条件下测试的 MoO₃ 和 Ir-MoO₃ 催化剂的表观活化能(Ea)。T 表示温度,r 表示 CO 生成速率。
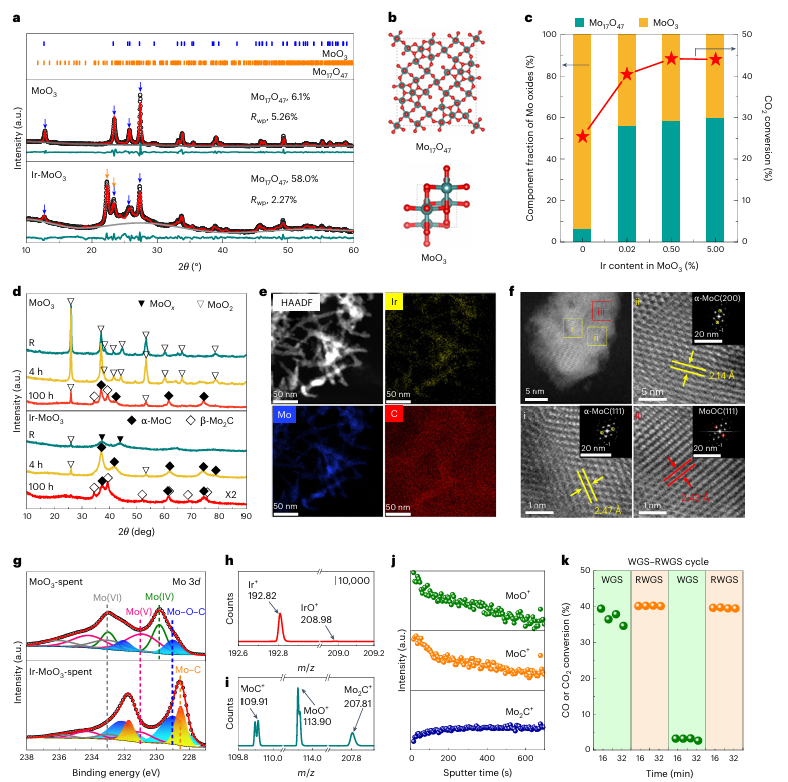
图2 | Mo物种的相和结构分析 a. MoO₃和Ir-MoO₃催化剂的精细XRD图谱。观察到的图谱用黑色表示,计算出的图谱用红色表示。MoO₃和Mo₁₇O₄₇的Bragg反射位置分别用蓝色和橙色刻度标注。2θ为透射光束与反射光束之间的角度。b. MoO₃和Mo₁₇O₄₇结构的超级晶胞视图,Mo原子用蓝色表示,O原子用红色表示。c. 不同MoO₃样品中的Mo氧化物成分比例及其在600°C下的RWGS反应中的催化性能。d. 还原400°C(R)后的MoO₃和Ir-MoO₃催化剂以及在600°C下4小时和100小时RWGS反应后的XRD图谱。e. Ir-MoO₃的高角环形暗场STEM图像及Ir、Mo、C元素的映射图。f. Ir-MoO₃的HAADF-STEM图像,插图为标记区域的快速傅里叶变换(FFT)图(i, ii和iii)。g. MoO₃和Ir-MoO₃催化剂在RWGS反应4小时后的Mo 3d XPS光谱(标记为MoO₃-spent和Ir-MoO₃-spent)。h–j. 通过飞行时间二次离子质谱(ToF-SIMS)对Ir (h)和Mo (i)物种的鉴定,以及它们在Ir-MoO₃-spent催化剂表面和次表面的分布(j)。m/z表示质荷比。k. Ir-MoO₃-spent催化剂的WGS-RWGS循环测试,以评估表面活性物种。WGS反应条件:0.1 MPa,200°C;5% CO,20% H₂O,He;GHSV = 60 l gcat⁻¹ h⁻¹。RWGS反应条件:0.1 MPa,600°C;24% CO₂,72% H₂,N₂;GHSV = 300 l gcat⁻¹ h⁻¹。
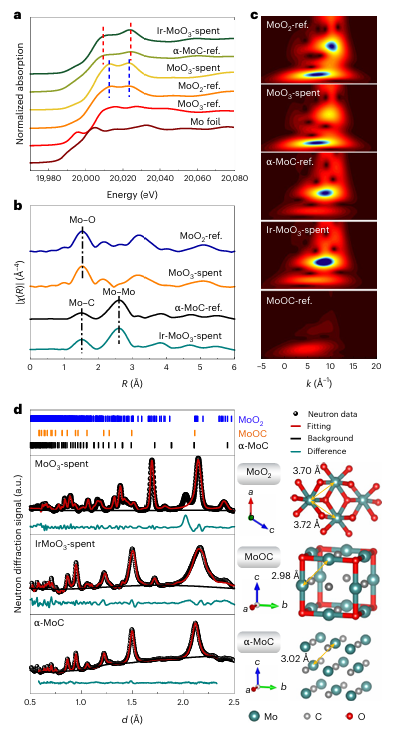
图3 | RWGS反应后MoO₃和Ir-MoO₃催化剂的结构表征 a. 归一化的XANES光谱;ref为参考样品,归一化XANES光谱的峰用虚线标出。 b. R空间中k³加权的EXAFS光谱(径向距离)。χ表示吸收截面。 c. Mo K边的EXAFS振荡小波变换图,显示MoO₃-spent、Ir-MoO₃-spent、Mo箔、MoO₂、MoO₃、α-MoC和MoOxCy参考样品的特征。 d. α-MoC、MoO₃-spent和Ir-MoO₃-spent样品的中子衍射(ND)图谱以及基于拟合结果的Mo物种模型。
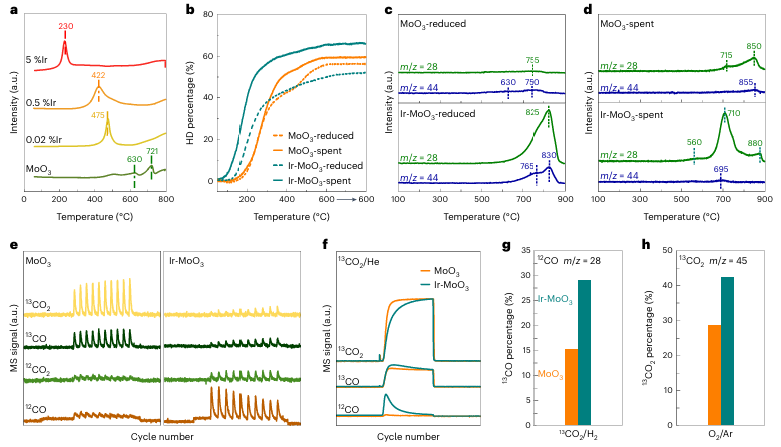
图4 | 反应物活化和活性位点调查 a. MoO₃及不同Ir掺杂量的MoO₃物种的H₂温度程序还原图谱。b. 还原样品(虚线)和使用后的样品(实线)的H/D交换百分比,箭头指示在600°C下保持的温度。c,d. 还原样品(c)和使用后样品(d)的CO₂温度程序脱附图谱,监测CO (m/z=28) 和CO₂ (m/z=44)信号。e,f. 在600°C下通过13CO₂同位素标记进行的脉冲(e)和突破(f)实验,针对MoO₃-spent和Ir-MoO₃-spent样品。g. 在13CO₂/H₂气氛下的突破实验中,生成的12CO百分比的比较。MS表示质谱仪。h. 在O₂/Ar气氛下的后续氧化处理中生成的13CO₂百分比。
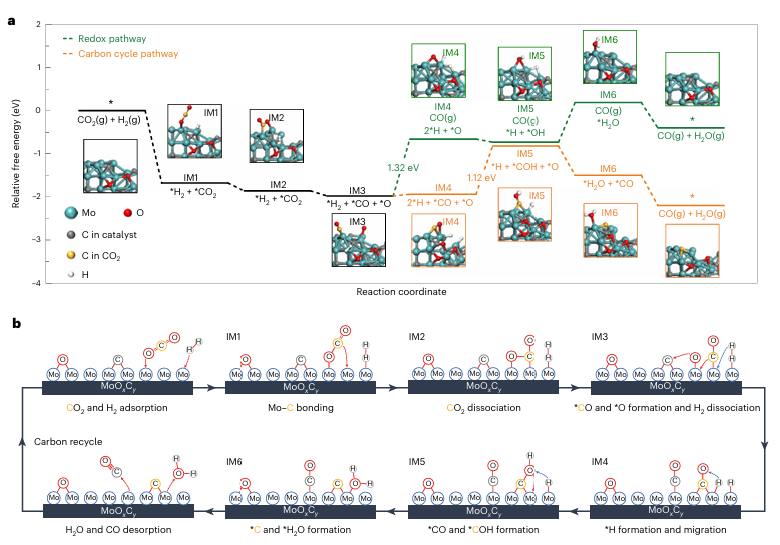
图5 | 通过DFT计算的RWGS反应机制 a. 基于MoOxCx模型的氧化还原路径(绿色)和碳循环路径(橙色)的能量剖面。每一步最有利的中间体(IM)在模拟中被标记,Mo、O、C和H原子分别用青色、红色、灰色和白色表示,CO₂中的C原子用黄色表示。 b. 碳循环路径的示意图,展示了从反应物吸附到产物脱附以及碳循环过程。
试验方法
1.X射线吸收近边结构(XANES)
方法说明:XANES可以用来分析元素的化学态和局部结构。
案例说明:XANES光谱用于确认Mo的价态变化,显示Mo在不同催化剂样品中的特征峰,表明Ir掺杂影响Mo物种的还原和碳化过程。
2.扩展X射线吸收精细结构(EXAFS)
方法说明:EXAFS可以提供邻近原子种类、数目和距离等信息,揭示材料的局部结构。
案例说明:EXAFS光谱进一步支持了Mo物种在反应前后局部结构的变化,表明Ir掺杂促进了MoC和Mo氧碳化物的形成过程。
3.X射线光电子能谱(XPS)
方法说明:XPS用于分析表面元素的化学组成及其化学态。
案例说明:XPS光谱揭示了催化剂在反应过程中的化学变化,提供了Mo物种的价态分布和氧空位信息。
4.高角环形暗场扫描透射电子显微镜(HAADF-STEM)
方法说明:HAADF-STEM可以直接观察纳米尺度的结构和元素分布。
案例说明:STEM图像清晰展示了α-MoC和Mo氧碳化物的形成,并揭示了Ir掺杂如何影响MoO₃结构的变化。
5.飞行时间二次离子质谱(ToF-SIMS)
方法说明:ToF-SIMS可以用来分析表面物种的分布及其质量-电荷比(m/z)。
案例说明:ToF-SIMS用于检测Ir-MoO₃催化剂的表面和次表面物种,包括MoO⁺、MoC⁺和Mo₂C⁺,以帮助理解碳循环路径
6.密度泛函理论(DFT)计算
方法说明:DFT计算用于模拟反应中的电子结构和反应路径,计算反应的能量剖面和活化能。
案例说明:DFT计算用于研究RWGS反应中的氧化还原路径和碳循环路径,揭示Mo氧碳化物表面CO₂还原过程中碳插入与CO生成的机制,进一步阐明Ir掺杂如何通过促进碳循环路径来提高催化剂的活性和稳定性。
▼▼▼
我们提供相关测试表征和理论计算服务,包括X射线吸收近边结构(XANES)、飞行时间二次离子质谱(ToF-SIMS)和密度泛函理论(DFT)计算等服务。如果您有相关需求,可以联系我们。
添加下方微信好友,立即咨询选择我们的科研服务,带你发顶刊走在化学领域的最前沿电话/微信:17621920434创新思路
本论文的创新点在于提出了一种基于Ir掺杂的Mo氧碳化物催化剂体系,通过火焰喷雾热解(FSP)法制备MoO₃催化剂,在反应过程中实现原位碳化,形成具有不饱和结构的Mo氧碳化物活性位点。该方法避免了传统高能耗的碳化预处理,并通过掺杂微量Ir,显著促进了催化剂的还原和碳化过程。此外,首次揭示了碳循环路径在逆水煤气变换反应中的重要性,提出了碳插入和氧空位生成机制,显著提高了催化剂的CO₂转化效率和稳定性。
总结展望
本研究成功开发了一种基于Ir掺杂Mo氧化物的高效CO₂转化催化剂,利用反应过程中原位形成的Mo氧碳化物活性位点,展现出优异的催化性能和长期稳定性。通过揭示碳循环路径及氧空位生成机制,进一步优化了逆水煤气变换反应(RWGS)的CO₂转化效率。该催化剂体系为高温CO₂转化应用提供了新的思路,特别是为工业规模的CO₂捕集与利用奠定了基础。进一步研究中,可以探索不同金属掺杂对催化剂性能的影响,并优化反应条件以提升其在实际工业环境中的适用性。
我是元素魔方科研服务
为多所高校、机构和企业提供专业科研支持
在节约科研成本的同时
还提供高效、全面的科研解决方案
我正好专业,你正好需要


















 313
313

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











