






来源公众号:分子动力学
文献解读
01
派言 写在前面
简化模型的定义是物理科学的核心;蛋白质也不例外。描述蛋白质折叠和动力学的统计力学方法,以及MD轨迹的分析,已经证明了在大型生物分子系统中,尽管组成整个系统的原子数以十万计,但缓慢的过程可以用更少的变量来描述。
本着这种精神,许多粗粒化(CG)模型被提出,通过MD和能量最小化来研究蛋白质。这些CG模型已被用于研究蛋白质折叠、分子间相互作用、蛋白质介导的膜现象的原理,并预测具有直接医学意义的新生物系统。
原子模型已经能够探索小球状蛋白质的相关景观;那么,是否存在一种分辨率使得化学可转移CG模型能够定量描述任意蛋白质的构型景观仍然是一个悬而未决的问题。
一个可转移的CG蛋白模型将产生重大影响。通过使用超级计算机或结合马尔可夫状态模型的分布式模拟,可以在毫秒时间尺度上模拟小分子溶剂化蛋白质的动力学。
近年来,使用机器学习(ML)方法进行分子模拟的兴趣激增,包括从大量数据中学习CG模型。从某种意义上说,ML CG模型的发展可以看作是正在进行的从量子力学计算中设计精确原子力场研究的延伸。
在这一领域,ML已经产生了高精度的力场,为突破性的计算研究提供了便利。尽管取得了这些进展,对于蛋白质或其他生物聚合物来说,完全自下而上的可转移CG模型仍然不存在。
可以说,粗粒化模型对于研究时间和长度尺度上的系统比原子模拟更为重要。而通过机器学习开发可迁移的粗粒化模型仍然面临着巨大的挑战。

鉴于此,德国柏林自由大学物理系Cecilia Clementi教授团队在《Current Opinion in Structural Biology》上发表研究论文。他们讨论了该领域的最新发展与面临的困难以及克服这些困难所做的努力。
CG模型主要是通过指定它们的分辨率来设计的,它定义了粗略的自由度,并通过它们的有效能量函数来说明这些珠子是如何相互作用的。
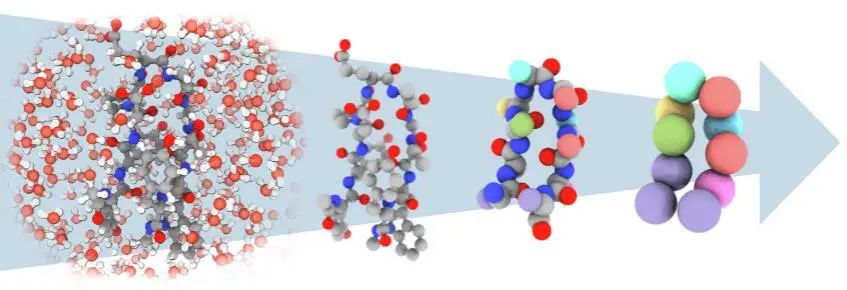
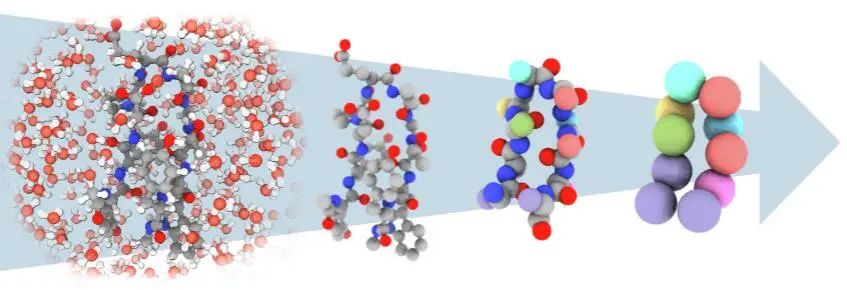
图1:从包全原子溶解表征到隐式的溶剂表征
传统方法上,选择分辨率首先是使用化学直觉或通过优化来重现所选择的属性。然后对模型的有效能量函数进行参数化以再现实验或模拟数据。
他们讨论的可迁移CG模型基本目标是预测不用于其参数化的蛋白质构象景观,理想情况下只使用感兴趣蛋白质的一级结构。
完整PDF原文后台回复“机器学习”获取。
02
解读 内容梳理
热力学一致性:为什么它很难?
自下而上的粗粒化通常模拟以下自由能面(U),称为有效CG能:
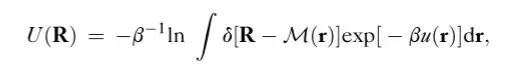
图2:公式(1)表达式
虽然热力学一致的CG能量由公式(1)唯一定义为常数。上述公式对于非平凡系统,积分无法求解因此,人们提出了多种近似U的策略。
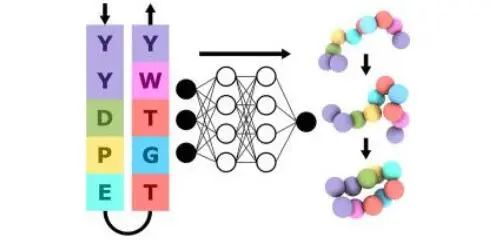
原则上,公式(1)表明,一旦选择了CG分辨率,CG模型的创建应该是简单的。然而,设计一个准确的ML CG模型并非易事(如图3所示)。

图3:从原子模拟数据和实验测量结果中创建和使用ML CG模型
1、训练CG力场的困难
用机器学习的力场进行自下而上粗粒化的主要挑战在于找到一个合适的ML公式,直接或间接地估计公式(1)中描述的难以解决的积分。
与从量子力学数据中学习原子势能面相比,这种情况更为困难,因为量子力学数据中的参考能量和力是已知的:
当学习CG自由能时,对于一个给定的CG结构R,U和它的梯度都不知道,因为公式(1)的积分是难以解决的。
2、CG表示法的选择
在设计原子力场时,玻恩-奥本海默近似法证明了电子自由度和核自由度之间的分离,并为有效的核势能表面提供了框架。
然而,对于CG模型来说,尺度的分离就不那么清楚了。因此,CG分辨率的选择非同小可,并影响到必须学习的自由能表面。
3、多体有效CG(自由)能量的函数形式
在实践中,通过平衡数据的力匹配训练ML CG模型需要一个基线(或 "先验")势,以减少灾难性的不正确推断到相空间的非物理区域。最终,一个好的先验势结合了物理原理,降低了学习的复杂性,并允许稳定的模拟。
因此,ML方法必须适应大的训练集,并且不应随着珠子类型的增加而增加,以便在考虑可转移模型时,评估时间不会增加。
在撰写本文时,最先进的ML CG模型可以定量地再现小蛋白质的行为。目前,描述较大蛋白质的最大障碍是收集足够的训练数据。
这种方法在多大程度上可以扩展到定义可转移的CG模型仍然是一个开放的问题。它可能只适用于一类蛋白质,或在特定的分辨率下。
在ML方法出现之前,这些问题仍然难以回答,因为原子学模型和CG模型之间的热力学一致性只能近似地执行;不清楚可转移模型的限制是由于CG能量的有限表达能力和有限的参考数据,还是由于可转移性的更基本问题。
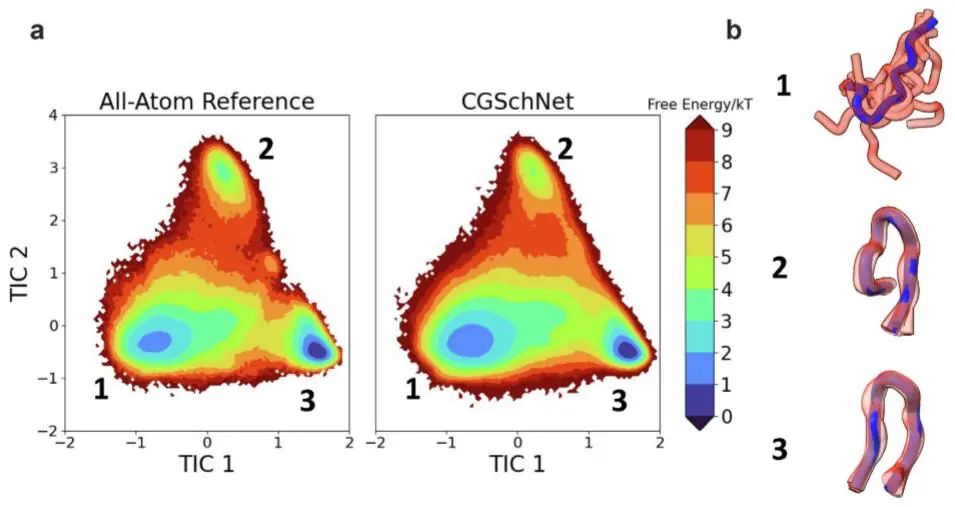
图4:基准蛋白CLN025上的Ca CG ML模型
现在,由于ML CG模型可以定量地执行单个蛋白质的热力学一致性,有工具来解决这些问题,并探索准确性和可转移性之间的权衡。在此,研究者讨论了实现这一目标的挑战,但他们仍然乐观地认为这样的研究方向是可以实现的。
03
总结 未来展望
即使一个可转移的自下而上的ML CG模型可以被定义,最终,计算模型的成功取决于它与实验的比较。自下而上的CG模型依赖于参考的原子学模型,必然会继承它们的不精确性和缺陷。
随着原子力场的改进,研究者期望CG模型也能变得更加精确。然而,即使是CG和原子学模型之间微小的不一致,也可能复合成与实验数据的重大偏差。
研究者相信,最终自下而上的ML CG将需要与自上而下的模型合并,以实现其有效和预测性的应用。
征稿启事
“分子动力学”自创号以来得到了广大科研人士的关注和支持。为更好地服务分子动力学研究和应用,本公众号现长期招聘供稿作者。
真诚欢迎MD相关科研爱好者加入“分子动力学”团队!可发送简历至likeapoem@163.com,邮件主题请注明“姓名+供稿作者”。















 9769
9769











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









