





muthor手册)
MiSeq SOP
准备
需要的文件
https://www.mothur.org/w/images/d/d6/MiSeqSOPData.zip
https://www.mothur.org/w/images/9/98/Silva.bacteria.zip
https://www.mothur.org/w/images/5/59/Trainset9_032012.pds.zip
下载好后,解压,将Trainset9_032012.pds 和silva.bacteria 文件夹移动到MiSeq_SOP文件夹下。
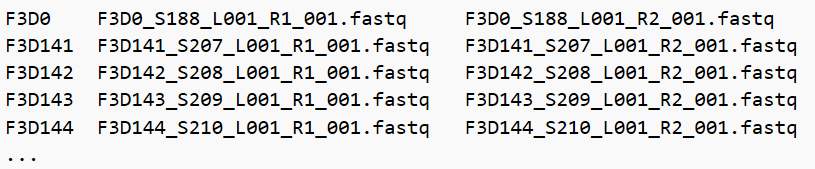
在MiSeq_SOP里,你可以看见22个fastq文件和 HMP_MOCK.v35.fasta
(我是将文件放在16sRNA文件夹下,如图)

Getting started
—创建一个stability.files
mothur > make.file(inputdir=fastq文件的路径, type=fastq, prefix=stability)
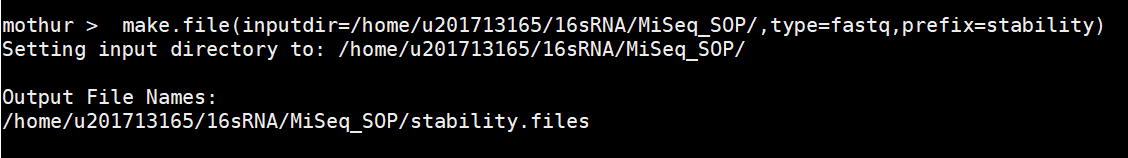
得到的stability.files应该是这样的:
第一列是样本名,第二列是正向读段,第三列是反向读段
reducing sequencing and PCR errors
我们要干的第一件事儿就是合并每个样本的2个读段,最后在合并样本数据。这可以通过make.contigs命令来实现(stability.files作为输入)。这个命令会提取fastq文件里的序列和质量分数、创建反向读段的反向互补,然后再把这些读段加入到重叠群里。这是一个非常简单的算法。首先,我们比对成对的序列,然后检查2条序列的每一个不匹配的位点。如果一条序列有碱基,另一条是空位,那么我们要求改位点的质量分数必须高于25才正确;如果2条序列在某个位点都有碱基,那么我们要求其中一个碱基的质量分数是6或者质量分数大于另一个碱基。
make.contigs(file=stability.files, processors=8)
你首先看到得是利用fastq文件产生了fasta和qual文件,然后制作重叠群(contigs)

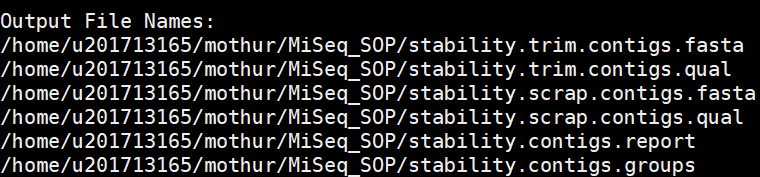
在最后它可能会出现:
[WARNING]: your sequence names contained ‘:’. I changed them to ‘_’ to avoid problems in your downstream analysis.
不要担心这个,序列的名字一般类似于"M00967:43:000000000-A3JHG:1:1101:18327:1699"这种。这就存在问题了,这个序列名太长了,而且序列名当中含有’ : ‘,这就会给后面构建进化树造成麻烦。所以我们就用’ - ‘代替了’ : '。这一命令会产生几个文件:
stability.trim.contigs.fasta and stability.contigs.groups
summary.seqs(fasta=stability.trim.contigs.fasta)

结果显示有152360条序列,其中大部分都是252~253bp。有趣得是,最长的是503bp,回想一下,这些序列应该全是251bp,那么这个读段显然不正确。而且,我们注意到,至少2.5%的序列存在不明确的碱基响应。
我们将使用screen.seqs来解决这个问题:
screen.seqs(fasta=stability.trim.contigs.fasta, group=stability.contigs.groups, maxambig=0, maxlength=275)
这条命令会去除含ambiguous bases和长度大于275的序列。还有一种实现方法:
screen.seqs(fasta=stability.trim.contigs.fasta, group=stability.contigs.groups, summary=stability.trim.contigs.summary, maxambig=0, maxlength=275)
这种方式可能会更快,因为summary.seqs输出文件有ambiguous bases和序列长度。此外。muthor非常聪明,记得我们在make.contigs的时候用的是8个处理器,所以一直会用8 processors
接下来,我们来看看muthor知道些什么:
get.current()

也就是说,mothur记得你最近使用的文件和processors。
你可以试着执行以下命令:
summary.seqs(fasta=stability.trim.contigs.good.fasta)
summary.seqs(fasta=current)
summary.seqs()
这时候,测序的错误率已经下降了一个数量级以上,我们现在也只有128872 条序列。
processing improved sequences
我们期望我们得到的很多序列都互为副本。由于比对多次相同的序列是在计算上是非常浪费,于是我们用unique.seqs()来去重。
unique.seqs(fasta=stability.trim.contigs.good.fasta)

如果2条序列相同,那么他们应该被合并成一个。看看屏幕输出,有2列数据,第一列是序列数,第二列是unique sequences。在unique.seqs()执行完之后,序列数量由128872减到16426。这就使我们的工作更简单了。为了更加简化,我们会制作一个表,行代表序列名称,列代表组的名字。
结果产生文件 stability.trim.contigs.good.count_table
.
summary.seqs(count=stability.trim.contigs.good.count_table)

是不是觉得很酷?现在我们就开始比对了。为了进程更容易,我们用pcr.seqs命令制作了一个数据库。要想运行这条命令,首先我们得有一个参考数据库((silva.bacteria.fasta),还必须知道在比对从哪里开始、哪里结束。将keepdots设置为false,去除前后的点。
pcr.seqs(fasta=silva.bacteria.fasta, start=11894, end=25319, keepdots=F, processors=8)

rename.file(input=silva.bacteria.pcr.fasta, new=silva.v4.fasta)

summary.seqs(fasta=silva.v4.fasta)

现在,我们有了定制的参考比对。参考比对有13425列(而非50000列),这大大减少了硬盘的空间,提升比对质量。
align.seqs(fasta=stability.trim.contigs.good.unique.fasta, reference=silva.v4.fasta)

summary.seqs(fasta=stability.trim.contigs.good.unique.align, count=stability.trim.contigs.good.count_table)

这是什么意思呢?可以看出,大多数序列在1968位点开始,11550位点结束。一些序列也在1250、1968开始,10693、13400结束。这些差异可能是由于在比对结束时的插入缺失。有时开始、结束位点都相同的序列可能比对结果不好,这是由于非特异性的扩增。为了确保没一条序列都重叠了相同部分,我们再次运行screen.seqs命令,得到start于位点1968或更靠前、end于位点11550甚至更后面。
screen.seqs(fasta=stability.trim.contigs.good.unique.align, count=stability.trim.contigs.good.count_table, summary=stability.trim.contigs.good.unique.summary, start=1968, end=11550, maxhomop=8)
summary.seqs(fasta=current, count=current)

现在我们知道我们的序列重叠的坐标,我们想确保序列只在特定的区域重复。所以我们会把不满足条件的序列过滤掉。由于配对测序已经完成,所以过滤不是难事儿。用filter.seqs来实现
filter.seqs(fasta=stability.trim.contigs.good.unique.good.align, vertical=T, trump=.)
运行结果

这意味着我们最初的队列是13425列,用trump=.和vertical=T.可以过滤掉13049个终端间隙字符和垂直间隙支付。最后的队列长度是376列。在修剪末端的时候,我们可能创造了一些冗余,所以运行unique.seqs
unique.seqs(fasta=stability.trim.contigs.good.unique.good.filter.fasta, count=stability.trim.contigs.good.good.count_table)

这已经识别出由之前的3条特有序列合并在一起的重复序列。接下来我们要预聚类,即去噪(允许出现2个错误)。这条命令将会对序列分组,按照丰度从高到低排列,识别出有2个差异的序列。如果序列被合并,我们倾向于允许100bp里有一个差异:
pre.cluster(fasta=stability.trim.contigs.good.unique.good.filter.unique.fasta, count=stability.trim.contigs.good.unique.good.filter.count_table, diffs=2)
现在我们有6087条序列。之前我们一直在除去测序错误,现在就要来删除嵌合体。采用VSEARCH算法,在mothur里,就是 chimera.vsearch命令。这个命令同样也会按照样本拆分数据、查找嵌合体。最好是有一个海量的序列作为参考。如果一条序列在样本中被标记为嵌合体,程序会默认将它从所有样本中删除。经验告诉我们,实际上很少的序列会被标记。操作如下:
chimera.vsearch(fasta=stability.trim.contigs.good.unique.good.filter.unique.precluster.fasta, count=stability.trim.contigs.good.unique.good.filter.unique.precluster.count_table, dereplicate=t

继续从fasta文件中取出嵌合体:
remove.seqs(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.fasta, accnos=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.accnos)


序列已经从128655减少到118171.这个序列数非常适合去标记嵌合体。作为控制质量的最后一步,需要判定数据集里面是否存在不满足期望的。继续用贝叶斯分类器:
classify.seqs(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.fasta, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.count_table, reference=/home/u201713165/mothur/MiSeq_SOP/trainset9_032012.pds.fasta, taxonomy=/home/u201713165/mothur/MiSeq_SOP/trainset9_032012.pds.tax, cutoff=80)
已经分好类了,现在开始删除吧remove.lineage(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.fasta, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.count_table, taxonomy=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.taxonomy, taxon=Chloroplast-Mitochondria-unknown-Archaea-Eukaryota)
Assessing error rates
只有在和mock共同排序时,才能测量错误率。首先用get.groups从mock中提取序列:
get.groups(count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.count_table, fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.fasta, groups=Mock)
结果显示,我们有64条unique序列,总共4048条。
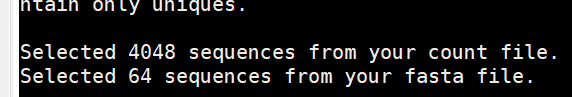
seq.error得到错误率
seq.error(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, reference=/home/u201713165/mothur/MiSeq_SOP/HMP_MOCK.v35.fasta, aligned=F)

错误率只有0.0065%。现在可以聚类到OTU,看看有多少假的OTU。
dist.seqs(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta, cutoff=0.03)
cluster(column=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.dist, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table)
make.shared(list=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.list, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, label=0.03)
rarefaction.single(shared=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.shared)
这串命令最后会生成一个文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.an.unique_list.groups.rarefaction
打开它,你可以看到,Mock样本离得4048条序列被聚类成35OTUs,这包含了一些隐藏的嵌合体。如果用3000条聚类,会生成31OTUs。在无嵌合体和测序错误的情况下,应当有20 OTUs。但事实并非这样,所以这个结果很好!
Preparing for analysis
现在有2件事儿做:为OTU、phylotypes分配序列。首先,从数据集里除去Mock:
remove.groups(count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.count_table, fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.fasta, taxonomy=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.taxonomy, groups=Mock)
OTUs
现在我们有几个选项可以将序列聚类到OTU中。
对于像这样的小数据集,我们可以使用dist.seqs和cluster来执行传统方法:
dist.seqs(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta, cutoff=0.03)
cluster(column=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.dist, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table)

也可以用另一种方法
用cluster.spilt命令。有点是更快,占内存更少。
cluster.split(fasta=stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta, count=stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, taxonomy=stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.taxonomy, splitmethod=classify, taxlevel=4, cutoff=0.03)
接下来,我们想知道每个OTU里面有多少条序列。用make.shared来实现,这里,有必要告诉你,mothur只对cutoff 0.03感兴趣.
make.shared(list=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.list, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, label=0.03)
我们或许也想只对每个OTUs的分类。classify.otu:
classify.otu(list=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.list, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, taxonomy=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.taxonomy, label=0.03)
打开stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.0.03.cons.taxonomy 你会看见:

这告诉我们Otu008 在样本里被观测到5337次,所有的序列(100%)都被分类到Alistipes.
Phylotypes
对于某些分析而言,可能希望将序列分成种系型。ptylotype:
phylotype(taxonomy=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.taxonomy)

phylotype的cutoff与cluster的有些不一样。相信你看到了屏幕上1~6的数字,分别对应着种到界。如果你想要属级别分享文件,那么如下操作:
The cutoff numbering is a bit different for phylotype compared to cluster/cluster.split. Here you see 1 through 6 listed; these correspond to Genus through Kingdom levels, respectively. So if you want the genus-level shared file we’ll do the following:
make.shared(list=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.tx.list, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, label=1)
classify.otu(list=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.tx.list, count=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table, taxonomy=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.taxonomy, label=1)
Phylogenetic
如果你对依赖于系统发育树和unifrac 命令的方法感兴趣,如计算系统发育多样性。那么就要构建系统发育进化树。下面就用dist.seqs和 clearcut.
dist.seqs(fasta=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta, output=lt, processors=8)
clearcut(phylip=/home/u201713165/mothur/MiSeq_SOP/stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.phylip.dist)
后面的数据分析,包括Alpha diversity、Beta diversity以后再翻译。














 2345
2345











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言










{kind=link}