







Mothur是一款由密歇根大学微生物与免疫学系的Patrick Schloss教授及其研究团队共同研发的微生物群落生态学分析工具,具有开源、可拓展的特点,能够满足微生物群落生态学的生物信息学分析要求。
Mothur整合了dotur、sons、treeclimber、s-libshuff、unifrac等功能,主要用于形成OTU/ASV,进行系统发育分析、计算Alpha和Beta多样性指数等。相较于其他生物信息学软件,Mothur具有命令简单、易上手和易理解的特点。
读者可以在Mothur官网https://mothur.org/下载其Windows版,并利用官网免费提供的标准化操作流程及命令说明,输入相关命令运行该软件,处理扩增子序列数据。目前,Mothur能够处理各种测序平台产生的序列数据,具体包括:454个焦磷酸测序,llumina公司的HiSeq和MiSeq, Sanger测序法,以及PacBio和IonTorrent等代表的三代测序技术。
01数据分析准备
硬件配置:笔记本电脑联想小新Pro-13ARE 2020。其中,处理器:AMD R7-4800U 8核16线程;硬盘:512GB SSD;操作系统:Windows 10家庭中文版;系统类型:64位操作系统, 基于x64的处理器
数据下载:本教程所使用的数据是Schloss实验室用Illumina的MiSeq平台通过配对末端读取(PE reads)生成的16S rRNA基因序列,所采用的方法是使用index reads,在单次运行中同时处理大量样本(384个),将其应用于生成这些库最新的湿实验wet-lab 标准操作程序中以便于用户学习Mothur。下载地址:https://mothur.org/wiki/miseq_sop/
需要下载的文件:
来自schloss实验室的示例数据
https://mothur.s3.us-east-2.amazonaws.com/wiki/miseqsopdata.zip
基于SILVA的细菌参考比对
https://mothur.s3.us-east-2.amazonaws.com/wiki/silva.bacteria.zip
RDP训练集的Mothur格式版本(v.9)
https://mothur.s3.us-east-2.amazonaws.com/wiki/trainset9_032012.pds.zip
Mothur可执行文件
https://github.com/mothur/mothur/releases/tag/v1.45.3(网页生成的Mothur可执行文件下载链接属于临时链接,很快失效。这里仅提供下载网页地址)
我们也提供了以上4个文件永久有效的百度网盘下载地址:https://pan.baidu.com/s/1m7X7gXrrv1O0KSyCBgV1lA 提取码:1234
温馨提示:推荐使用电脑桌面端浏览器跳转下载!

注意:1.文件解压后,将Trainset9_032012.pds、silva.bacteria文件夹和Mothur可执行文件夹的内容移动到MiSeq_SOP文件夹中;
2.本实验是关于肠道微生物组的正常变化对宿主健康的影响。该实验欲探究的问题是:与第140天-150天检测到的微生物组相比,小鼠断奶后的前10天体重的快速增长是否影响了其微生物组的稳定性。分析的过程中将综合利用操作分类单元(OTU)、扩增子/精确序列变体(ASV/ESV)、phylotype和phylogenetic的组合来解决想要探究的问题。示例数据所包含的数据为:单只小鼠10个时间点(5个早期和5个晚期)的数据文件(the flow files),以及21株细菌的基因组DNA组成的模拟群体(Mock community)二次测序数据。
数据清单文件生成
MiSeq_SOP文件夹中的文件包含22个fastq文件,代表来自雌3和“1”模拟群落的10个时间点。其中HMP_MOCK.v35.fasta文件是以fasta格式排序的模拟群落中使用的序列。
最后有一个名为stability.files的文件,该文件的第一行如下所示:F3D0_S188_L001_R1_001.fastq和F3D0_S188_L001_R2_001.fastq,分别对应于第0天(即断奶当天)的雌性3。这些序列为250 bp,在16S rRNA基因的V4区overlap,该区域长约253 bp。
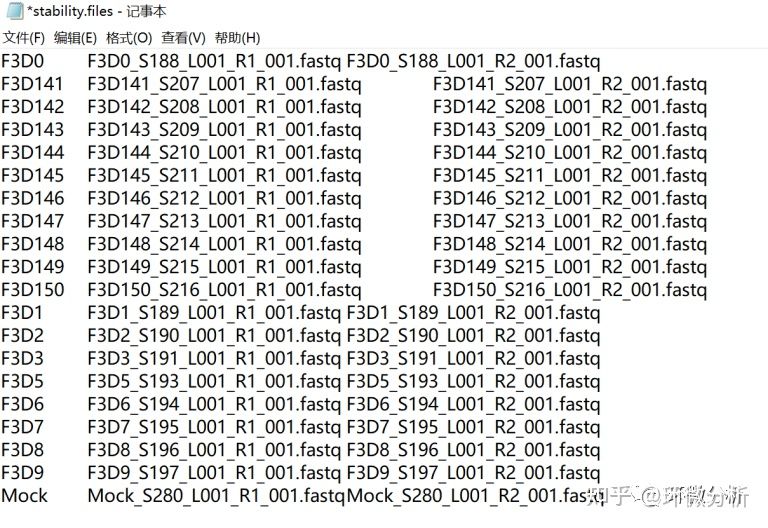
在Mothur中使用make.file命令创建此文件:
| 命令注释:make.file命令为序列的拼接(make.contigs)、创建输入文件。这个命令将多个文件连接起来并把结果输出到一个清单文件(stability.file)中。命令注释:make.file命令为序列的拼接(make.contigs)、创建输入文件。这个命令将多个文件连接起来并把结果输出到一个清单文件(stability.file)中。 |

输出文件:stability.file
| stability.file:清单文件。第一列是样本的名称,第二列是该样本的正向读取名称,第三列是该样本的反向读取名称。可以使用make.file命令创建,也可以手动编辑:可以先新建文本文档(TXT),按照stability.files文件模板写好后,修改文件后缀为.files即可。 |
02知识小便签
序列与测序
高通量测序技术(High-throughput sequencing):又称“下一代”测序技术("Next-generation" sequencing technology,NGS),是基于PCR和基因芯片发展而来的DNA测序技术。以能一次并行对几十万到几百万条DNA分子进行序列测定和一般读长较短等为标志。目前有很多用于微生物群落多样性研究的高通量测序平台。
Illumina MiSeq:2011年,Illumina推出台式测序平台MiSeq,具有体积和通量小、价格低的特点。MiSeq便于扩增子和细菌样本测序,测序原理为四通道可逆性末端边合成边测序技术。读长:2×300bp;reads:5×106;通量:15GB;样本数量:在单次运行中可对96个样品进行多重分析。
16S rRNA:指原核微生物(细菌和古菌)染色体上编码核糖体(Ribosome)RNA的16S(S是沉降系数)小亚基(Subunit)的基因序列。16S rRNA基因普遍存在于细菌和古细菌中,具有多个拷贝数,全长1500 bp左右,其结构由9个可变区(Variable region)和10个保守区(Conserved region)交替组成。保守区有利于扩增引物的设计,可变区体现了物种间的进化差异。对16S rDNA某个可变区进行测序,可以用于研究环境微生物中细菌或古菌的群落结构多样性。
18S rRNA:编码真核生物核糖体小亚基的DNA序列,其中既有保守区,也有可变区(V1−V9)。
ITS:16S rRNA和23S rRNA基因间的转录间隔区,是编码核糖体RNA前体基因的一段,位于核仁区,多用来进行进化分析。
Index reads:为了区分两个方向的reads,其中一个测序引物前面要添加一小段index序列进行标记,具体来说就是加到Illumina测序接头上的,保证多个测序文库可以在同一个flow-cell上或者同一个lane上进行混合测序(multiplexed)。
湿实验标准操作程序(wet-lab SOP):高通量测序实验室会购买核酸提取试剂盒、文库准备试剂盒、通用测序反应试剂盒等,或者自行设计富集区域,合成相应的引物探针等并且建立生物信息学分析流程。
干实验标准操作程序:“干实验”过程是高通量测序的一大特点。该过程由分析软件和算法组成,并且按照一定的流程进行。
扩增子结构

Barcode:样品标签,用于混池测序后区分序列来自哪个样本;
Primer:在16S/ITS/18S保守区设计的引物,用于扩增rRNA的部分高变区;
Amplicon:扩增的部分rRNA。
扩增子测序:对特定长度的PCR产物或者捕获的片段进行测序,分析序列中的变异。其原理是利用PCR的引物来扩增基因组的特定区域,靶向地捕获目标区域的DNA,达到目的DNA片段的富集目标,最后针对扩增产物(也被称为扩增子)进行高通量测序,分析序列中的遗传变异等信息的。
操作分类单元(OTU):在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。
数据库介绍
Silva数据库:SILVA一词起源于拉丁文silva(意为forest),它是一个包含三域微生物(细菌、古菌、真核)rRNA基因序列的综合数据库,其数据库涵盖了原核和真核微生物的小亚基rRNA基因序列(简称SSU,即16S和18SrRNA)和大亚基rRNA基因序列(简称LSU,即23S和28SrRNA)。因为SILVA数据库更新比较及时,因此是目前rRNA基因高通量测序后最常选用的参考数据库之一。此外,与RDP类似,SILVA也可被用于平时菌种鉴定时,对少量rRNA基因测序后的物种进行分类鉴定,此时主要用其SINA Alignment Service功能,可非常方便地确定某条rRNA基因序列从门到属/种水平的分类信息并给出各分类水平相应的置信度。
RDP数据库:RDP数据库全称“RibosomalDatabase Project”,该数据库提供质控、比对、注释的细菌、古菌16S rRNA基因和真菌28S rRNA基因序列。RDP是目前较常用的rRNA基因高通量测序后作为比对、注释的参考数据库。此外,还可用于平时菌种鉴定时,对少量rRNA基因测序后的物种进行分类鉴定,此时主要用其Classifier功能,可以非常方便地确定某条rRNA基因序列从门到属/种水平的分类信息并给出各水平相应的置信度。
Greengenes数据库:Greengenes是专门针对细菌、古菌16S rRNA基因的数据库,相比前面提到的RDP和SILVA数据库,该数据库更新速度较慢,目前更新停留在2013年5月更新的gg_13_5版本,目前较常用于16S rRNA基因高通量测序后进行嵌合体去除的参比数据库。
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!















 918
918











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









