






代码:
import os
def fasta_to_phy(fasta_file, phy_file):
seq_names = []
sequences = []
with open(fasta_file, "r") as f_in:
for line in f_in:
line = line.strip()
if line.startswith(">"):
seq_name = line[1:]
seq_names.append(seq_name)
sequences.append("")
else:
sequences[-1] += line
with open(phy_file, "w") as f_out:
f_out.write(f"{len(seq_names)} {len(sequences[0])}\n")
for i in range(len(seq_names)):
f_out.write(f"{seq_names[i]} {sequences[i]}\n")
print(f"Successfully converted {fasta_file} to {phy_file}.")
# 设置工作目录
os.chdir(r"/path/to/working/directory") # 将路径替换为工作目录路径
fasta_file = "input.fasta" # 输入的FASTA文件名
phy_file = "output.phy" # 输出的PHYLIP文件名
fasta_to_phy(fasta_file, phy_file)
fasta格式输入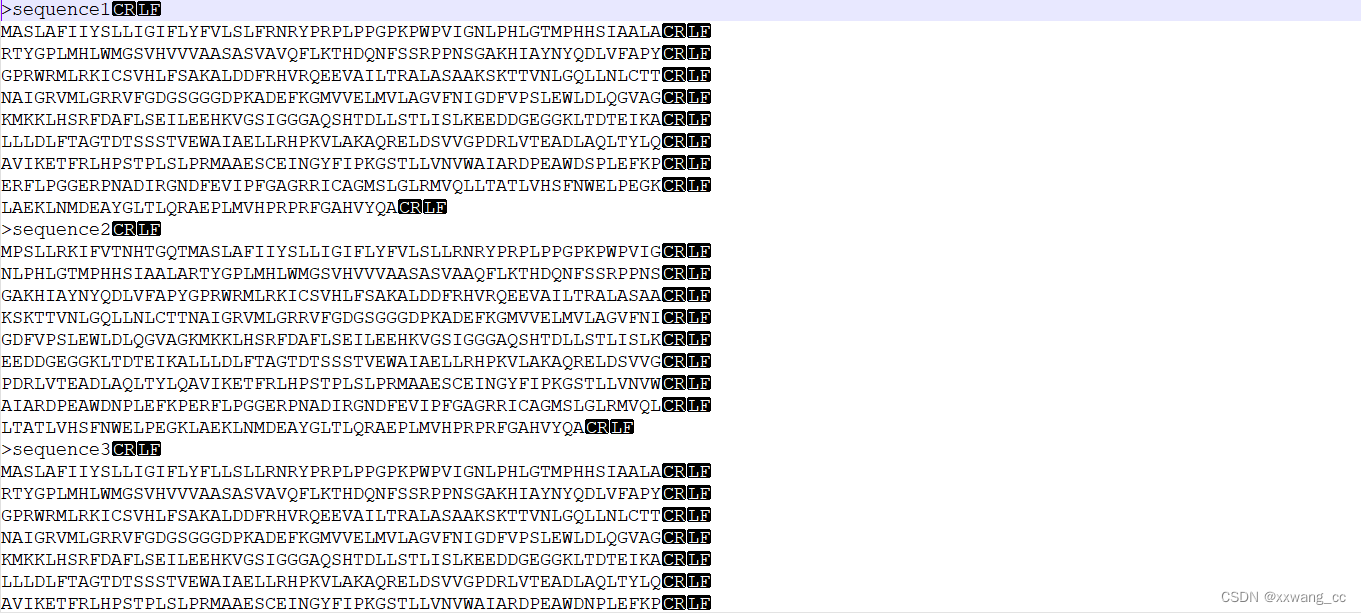
phy格式输出
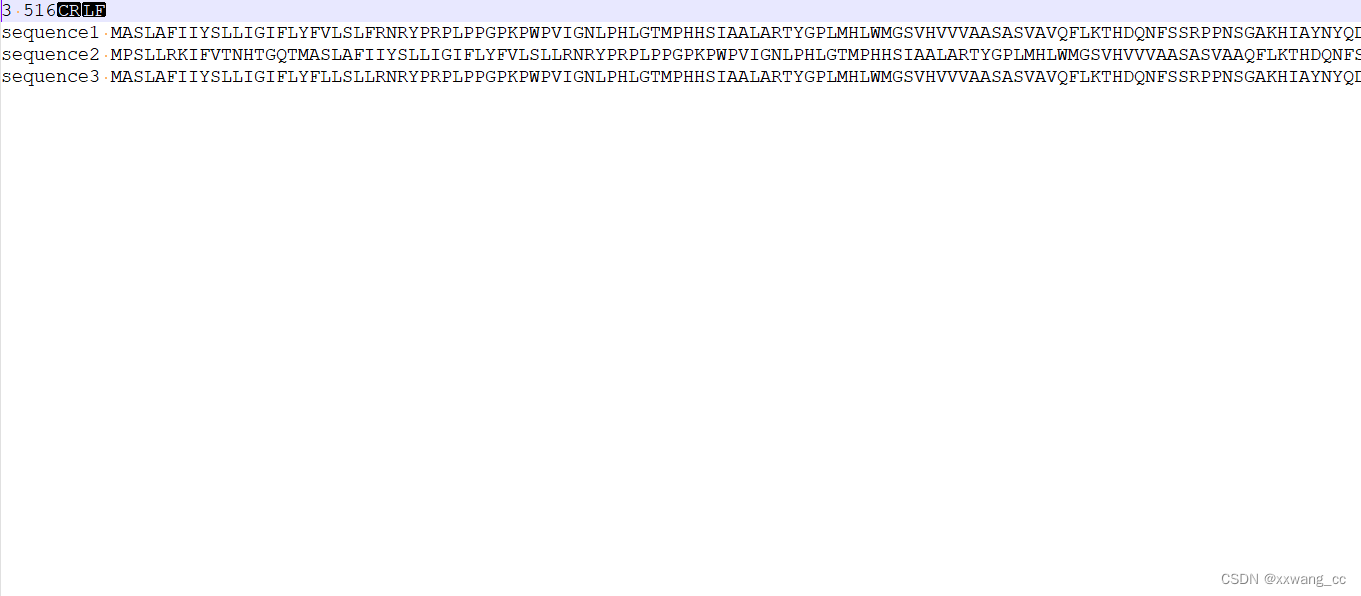















 2639
2639











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









