










MOSES+:Molecular Sets,A Benchmarking Platform for Molecular Generation Models
最主要的是看分子生成的评价指标。现有新的生成方法有很多,论文中的方式有点老了。
一、INTRODUCTION
虽然生成模型有很多,但如何对它们进行比较和排序尚不清楚。在这项工作中,引入MOSES的基准测试平台来标准化分子生成模型的训练和比较。MOSES提供训练和测试数据集,以及一套用于评估生成结构的质量和多样性的指标。
药物敏感分子的数量在10^23到10^80个化合物之间

1、Distribution Learning
给定一组训练样本Xtr={xtr1,…,xtrN},从未知分布p(x),分布学习模型近似于p(x)和某些分布q(x)。
药物化学家可能期望某些亚结构在生成结构中更普遍。依靠一组手动或自动选择的化合物,分布学习模型产生一个更大的数据集,保留数据集中的隐式规则。分布学习模型的另一个应用是扩展下游半监督预测任务的训练集:可以通过从生成模型中抽样化合物来添加新的未标记数据。
Distribution Learning model是度量p(x)和q(x)之间的偏差,该模型可以隐式或显式地定义probability mass function q(x)。显式模型,如隐马尔可夫(Hidden Markov)模型、n-gram language模型或normalizing flows,可以解析计算q(x)并从中采样。隐式模型,如VAE、adversarial autoencoder或generative adversarial networks可以从q(x)中采样,但无法计算出probability mass function q(x)的确切值。为比较这两种模型,考虑的评价指标仅依赖于q(x)中的样本。
2、Molecular Representations
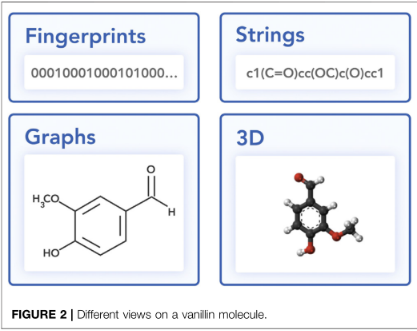
Strings:SMILES、DeepSMILES
Graph:Molecular graphs
3、Metrics
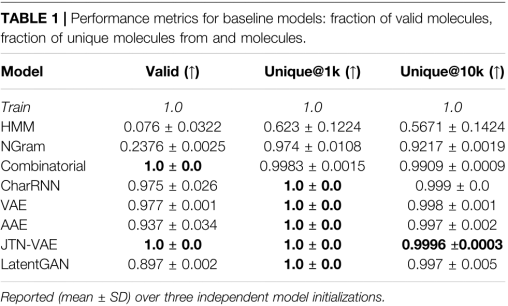
1)Fraction of valid (Valid) and unique (Unique@k)
生成的SMILES字符串的有效性和唯一性。K一般取1000或者10000,如果有效分子的数量小于K,则计算所有有效分子的唯一性。
Uniqueness检查模型是否崩溃(只产生几个典型分子),
2)Novelty
生成新分子中没有出现在训练集中的部分。低Novelty表明过拟合。
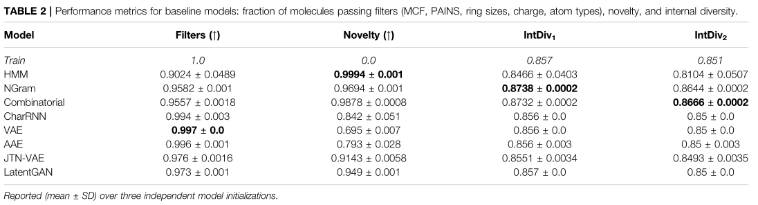
3)Filters
在数据集构建过程中通过filter的生成分子的比例。虽然生成的分子通常在化学上是有效的,但它们可能包含不需要的片段:在构建训练数据集时,我们删除了带有此类片段的分子,并期望模型避免产生它们。
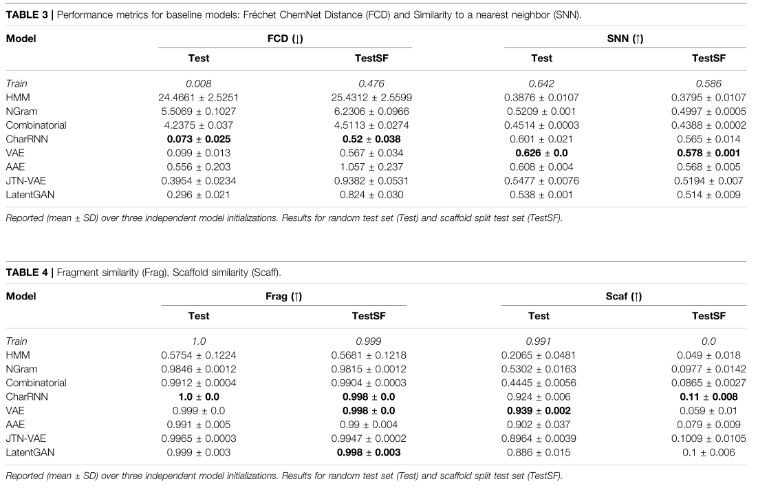
4)Fragment similarity (Frag)
比较BRICS片段在生成集和reference集中的分布。
表示cf (A)表示子结构f在集合A的分子中出现的次数,表示在G或R中出现的片段集为f,Frag metric用余弦相似度计算:
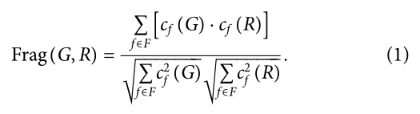
如果两组分子有相似的片段,则Frag metric较大。如果某些片段在生成集中表示过多或不足(或从未出现),则度量值将较低。这个度量的极限是[0,1]。
5)Scaffold similarity (Scaff)
比较frequencies of Bemis–Murcko scaffolds ,包含所有分子的环结构和连接环的连接子片段。使用RDKit,额外考虑碳基(carbonyl groups)连接到环作为scaffolds 的一部分。用cs(A)表示scaffolds s在集合A中的分子中出现的次数,用G或R中出现的片段集表示s,metric定义为余弦相似度:
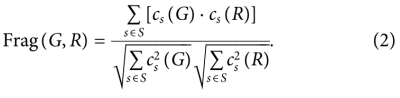
目的是显示生成数据集和reference集中的支架有多相似,范围为:[0,1]。
片段和支架的相似性都是在子结构水平上比较分子的。因此,即使G和R包含不同的分子,也有可能有一个相似的分子。
6)Similarity to a nearest neighbor (SNN)
生成集G中分子mG的指纹与reference数据集R中nearest neighbor分子mR的平均Tanimoto similarity T(mG,mR)(也称为Jaccard指数):
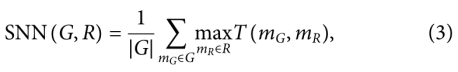
使用了标准的Morgan(扩展连接)指纹,其radius为2,RDKit计算1024位,范围为:[0,1]
7)Internal diversity (IntDivp)
评估生成的G分子集合内的化学多样性。
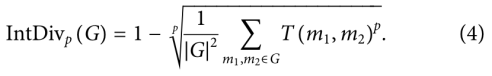
该指标检测生成模型是否——模式崩溃。模式坍塌后,产生的样本种类有限,忽略了化学空间的某些区域。该指标的值越高,表示生成集合的多样性越高。
该指标的值越高,表示生成集的多样性越高。metrix范围为[0,1]。
8)Fréchet ChemNet Distance (FCD)
通过训练用来预测药物生物活性的深度神经网络ChemNet倒数第二层的activations来计算的。计算分子的canonical SMILES表示的activations。activations捕获化合物的化学和生物特性。对于两组分子G和R, FCD定义为:
![]()
其中,μG、μR为均值向量,ΣG、ΣR为G集和R集分子活化的全协方差矩阵。FCD与其他指标相关。例如,如果生成的结构不够多样化(低IntDivp)或模型产生太多重复(低uniqueness),FCD将减少,因为方差更小。
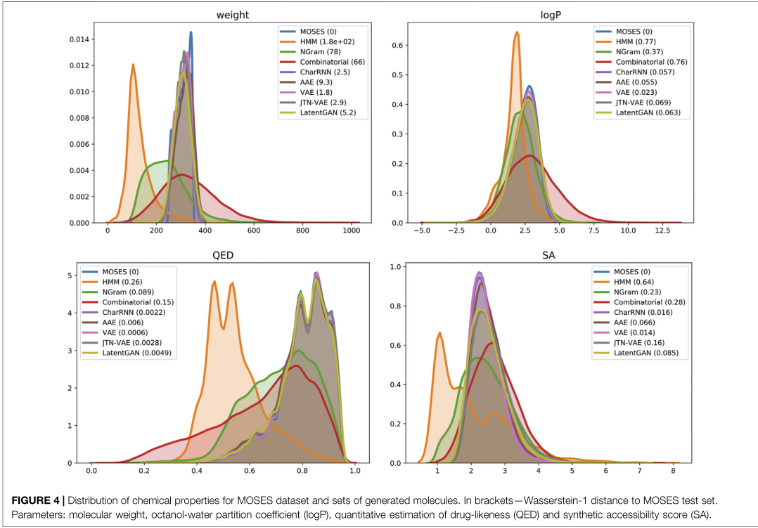
9)Properties distribution
属性分布是可视化评估生成结构的有用工具。为了定量比较生成集和测试集的分布,计算生成集和测试集的属性分布之间的1D Wasserstein-1距离
分子量(MW):分子中原子量的总和。通过绘制生成集和测试集的分子量直方图,可以判断生成集是偏向于较轻的分子还是较重的分子。
LogP:辛醇-水分配系数,是辛醇/水两相体系中化学物质在辛醇相中的浓度与在水相中的浓度之比;
合成可达性评分(SA):对合成一个给定分子的难易程度(10)或易易程度(1)的启发式估计。
药物相似度定量估计(QED):一个[0,1]值,用来估计一个分子成为药物可行候选分子的可能性。
二、DATASET
ZINC Clean Leads (包含45912,276个分子),分子量在250到350 Da之间,旋转键数不大于7,XlogP 不大于3.5。Clean-leads数据集由适合识别目标化合物的结构组成,它们足够小,可以对生成的分子进行进一步的ADMET优化。去掉了含有带电原子的分子,除C, N, S, O, F, Cl, Br, H外的原子,或大于8个原子的循环(代码在后面)。
分成三部分:训练(1,584,664个分子),测试(176,075个分子)和骨架测试(176,226个分子)
三、BASELINES
character-level recurrent neural networks (CharRNN) 、Variational Autoencoders (VAE) , Adversarial Autoencoders (AAE) 、Junction Tree Variational Autoencoders (JTN-VAE) , LatentGAN , and non-neural baselines(n-gram generative model, Hidden Markov Model (HMM), and acombinatorial generator)
数据处理代码:
"""Functions that can be used to preprocess SMILES sequnces in the form used in the publication."""
import numpy as np
import pandas as pd
import tensorflow as tf
from rdkit.Chem.SaltRemover import SaltRemover
from rdkit import Chem
from rdkit.Chem import Descriptors
REMOVER = SaltRemover()
ORGANIC_ATOM_SET = set([5, 6, 7, 8, 9, 15, 16, 17, 35, 53])
def dataframe_to_tfrecord(df,
tfrecord_file_name,
random_smiles_key=None,
canonical_smiles_key=None,
inchi_key=None,
mol_feature_keys=None,
shuffle_first=False):
"""Function to create a tf-record file to train the tranlation model from a pandas dataframe.
Args:
df: Dataframe with the sequnce representations of the molecules.
tfrecord_file_name: Name/Path of the file to write the tf-record file to.
random_smiles_key: header of the dataframe row which holds the randomized SMILES sequnces.
canonical_smiles_key: header of the dataframe row which holds the canonicalized SMILES
sequnces.
inchi_key: header of the dataframe row which holds the InChI sequnces.
mol_feature_keys:header of the dataframe row which holds molecualar features.
shuffle_first: Defines if dataframe is shuffled first before writing to tf-record file.
Returns:
None
"""
writer = tf.python_io.TFRecordWriter(tfrecord_file_name)
if shuffle_first:
df = df.sample(frac=1).reset_index(drop=True)
for index, row in df.iterrows():
feature_dict = {}
if canonical_smiles_key is not None:
canonical_smiles = row[canonical_smiles_key].encode("ascii")
feature_dict["canonical_smiles"] = tf.train.Feature(
bytes_list=tf.train.BytesList(value=[canonical_smiles])
)
if random_smiles_key is not None:
random_smiles = row[random_smiles_key].encode("ascii")
feature_dict["random_smiles"] = tf.train.Feature(
bytes_list=tf.train.BytesList(value=[random_smiles])
)
if inchi_key is not None:
inchi = row[inchi_key].encode("ascii")
feature_dict["inchi"] = tf.train.Feature(
bytes_list=tf.train.BytesList(value=[inchi])
)
if mol_feature_keys is not None:
mol_features = row[mol_feature_keys].values.astype(np.float32)
feature_dict["mol_features"] = tf.train.Feature(
float_list=tf.train.FloatList(value=mol_features)
)
example = tf.train.Example(features=tf.train.Features(feature=feature_dict))
serialized = example.SerializeToString()
writer.write(serialized)
writer.close()
def randomize_smile(sml):
"""Function that randomizes a SMILES sequnce. This was adapted from the
implemetation of E. Bjerrum 2017, SMILES Enumeration as Data Augmentation
for Neural Network Modeling of Molecules.
Args:
sml: SMILES sequnce to randomize.
Return:
randomized SMILES sequnce or
nan if SMILES is not interpretable.
"""
try:
m = Chem.MolFromSmiles(sml)
ans = list(range(m.GetNumAtoms()))
np.random.shuffle(ans)
nm = Chem.RenumberAtoms(m, ans)
return Chem.MolToSmiles(nm, canonical=False)
except:
return float('nan')
def canonical_smile(sml):
"""Helper Function that returns the RDKit canonical SMILES for a input SMILES sequnce.
Args:
sml: SMILES sequence.
Returns:
canonical SMILES sequnce."""
return Chem.MolToSmiles(sml, canonical=True)
def keep_largest_fragment(sml):
"""Function that returns the SMILES sequence of the largest fragment for a input
SMILES sequnce.
Args:
sml: SMILES sequence.
Returns:
canonical SMILES sequnce of the largest fragment.
"""
mol_frags = Chem.GetMolFrags(Chem.MolFromSmiles(sml), asMols=True)
largest_mol = None
largest_mol_size = 0
for mol in mol_frags:
size = mol.GetNumAtoms()
if size > largest_mol_size:
largest_mol = mol
largest_mol_size = size
return Chem.MolToSmiles(largest_mol)
def remove_salt_stereo(sml, remover):
"""Function that strips salts and removes stereochemistry information from a SMILES.
Args:
sml: SMILES sequence.
remover: RDKit's SaltRemover object.
Returns:
canonical SMILES sequnce without salts and stereochemistry information.
"""
try:
sml = Chem.MolToSmiles(remover.StripMol(Chem.MolFromSmiles(sml),
dontRemoveEverything=True),
isomericSmiles=False)
if "." in sml:
sml = keep_largest_fragment(sml)
except:
sml = np.float("nan")
return(sml)
def organic_filter(sml):
"""Function that filters for organic molecules.
Args:
sml: SMILES sequence.
Returns:
True if sml can be interpreted by RDKit and is organic.
False if sml cannot interpreted by RDKIT or is inorganic.
"""
try:
m = Chem.MolFromSmiles(sml)
atom_num_list = [atom.GetAtomicNum() for atom in m.GetAtoms()]
is_organic = (set(atom_num_list) <= ORGANIC_ATOM_SET)
if is_organic:
return True
else:
return False
except:
return False
def filter_smiles(sml):
try:
m = Chem.MolFromSmiles(sml)
logp = Descriptors.MolLogP(m)
mol_weight = Descriptors.MolWt(m)
num_heavy_atoms = Descriptors.HeavyAtomCount(m)
atom_num_list = [atom.GetAtomicNum() for atom in m.GetAtoms()]
is_organic = set(atom_num_list) <= ORGANIC_ATOM_SET
if ((logp > -5) & (logp < 7) &
(mol_weight > 12) & (mol_weight < 600) &
(num_heavy_atoms > 3) & (num_heavy_atoms < 50) &
is_organic ):
return Chem.MolToSmiles(m)
else:
return float('nan')
except:
return float('nan')
def get_descriptors(sml):
try:
m = Chem.MolFromSmiles(sml)
descriptor_list = []
descriptor_list.append(Descriptors.MolLogP(m))
descriptor_list.append(Descriptors.MolMR(m)) #ok
descriptor_list.append(Descriptors.BalabanJ(m))
descriptor_list.append(Descriptors.NumHAcceptors(m)) #ok
descriptor_list.append(Descriptors.NumHDonors(m)) #ok
descriptor_list.append(Descriptors.NumValenceElectrons(m))
descriptor_list.append(Descriptors.TPSA(m)) # nice
return descriptor_list
except:
return [np.float("nan")] * 7
def create_feature_df(smiles_df):
temp = list(zip(*smiles_df['canonical_smiles'].map(get_descriptors)))
columns = ["MolLogP", "MolMR", "BalabanJ", "NumHAcceptors", "NumHDonors", "NumValenceElectrons", "TPSA"]
df = pd.DataFrame(columns=columns)
for i, c in enumerate(columns):
df.loc[:, c] = temp[i]
df = (df - df.mean(axis=0, numeric_only=True)) / df.std(axis=0, numeric_only=True)
df = smiles_df.join(df)
return df
def preprocess_smiles(sml):
"""Function that preprocesses a SMILES string such that it is in the same format as
the translation model was trained on. It removes salts and stereochemistry from the
SMILES sequnce. If the sequnce correspond to an inorganic molecule or cannot be
interpreted by RDKit nan is returned.
Args:
sml: SMILES sequence.
Returns:
preprocessd SMILES sequnces or nan.
"""
new_sml = remove_salt_stereo(sml, REMOVER)
new_sml = filter_smiles(new_sml)
return new_sml
def preprocess_list(smiles):
df = pd.DataFrame(smiles)
df["canonical_smiles"] = df[0].map(preprocess_smiles)
df = df.drop([0], axis=1)
df = df.dropna(subset=["canonical_smiles"])
df = df.reset_index(drop=True)
df["random_smiles"] = df["canonical_smiles"].map(randomize_smile)
df = create_feature_df(df)
return df
















 3996
3996

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









