







电子生物?GenSLMs大模型探索SARS病毒的遗传变异
研究电子生命,做赛博科研:今天给大家介绍的文献是来自芝加哥大学、哈佛大学、加州理工学院、美国能源部下的Argonne National Laboratory、NVIDIA Inc、Cerebras Inc.共同集合了跨学科、跨领域的强大科研力量,通过大语言模型对SARS-CoV-2进化动态的研究。

文章内容梳理
摘要简介
背景是什么 RNA病毒(如SARS-CoV-2)在宿主感染期间高频率突变和快速适应的能力。监测和解析这些变异对于追踪疫情传播、评估病毒传染性、致病性及免疫逃逸能力至关重要。
做了什么? GenSLMs通过预训练于超过1.1亿个原核生物基因序列,并针对150万个SARS-CoV-2基因组进行精细调整,展现出能够快速、准确识别关注变异株(Variants of Concern, VoCs)的能力。
意义是什么? 这种模型具有泛化能力,有望应用于其他预测任务,标志着在病毒新变种识别与分类方法上的革新。
文章结果速览
一、模型总览

GenSLMs定位为超越传统蛋白质语言模型(PLMs),专门用于描述SARS-CoV-2进化动态的工具。不再局限于单一蛋白质的研究,而是利用全基因组数据来建模单个核苷酸级别的突变,间接考虑了在密码子级别上的蛋白质突变。图1展示了GenSLMs如何将SARS-CoV-2基因组的核苷酸序列作为输入,学习单个密码子的语义嵌入,并将这些嵌入转化为病毒编码的29个独立蛋白质序列。
但是,在基因组上训练大模型,有以下局限:
-
1.基因组全长包含约30,000个核苷酸(对应约10,000个密码子/氨基酸),LLMs在处理长序列时可能会过度关注局部而非全局模式;
-
2.SARS-CoV-2基因组之间整体相似度极高(>约99%),仅少数变异导致显著表型差异,需要模型能够有效捕获这些关键变化。
为了克服这些困难 GenSLM隐式识别了通过DNA转录和mRNA翻译产生个体蛋白质的内在逻辑层次(基于中心法则)。通过对来自BV-BRC的基因水平数据进行训练,以模拟GenSLMs的这一过程。 同时,虽然多个密码子可能编码相同的氨基酸,但是作者相信通过大力出奇迹,可以让模型自己学会这些东西。模型的框架使用的是这篇文章的框架:

二、数据搜集
这篇文章的data collection环节主要涵盖了两个关键数据源的获取和处理,旨在为构建GenSLMs(基因组尺度语言模型)提供高质量的SARS-CoV-2基因组数据和丰富的原核生物基因数据,以支持模型学习和识别病毒变异。
-
SARS-CoV-2基因组数据集
-
数据来源:利用了由Bacterial and Viral Bioinformatics Resource Center (BV-BRC)提供的超过150万条高质量、完整的SARS-CoV-2基因组序列。
-
数据筛选与质量控制:剔除了长度不足29,000 bp或含超过1%不确定碱基(ambiguous bases)的基因组序列,确保纳入的数据具有足够的完整性与准确性。
-
补充数据:研究团队还引入了来自休斯顿卫理公会医院系统(Houston Methodist Hospital System)的大规模SARS-CoV-2基因组数据。该数据集包含从2020年5月15日至2022年1月14日期间采集的约70,000份患者样本。
-
进一步的质量控制,移除序列两端各100个核苷酸以及Spike蛋白编码区域中因引物结合不良导致深度较低的56个位点,并丢弃含有超过256个不确定字符的序列。 最终筛选得到16,545条高质量序列,用于后续的基因组尺度系统发育分析。
-
Prokaryotic基因数据集
-
数据来源:为了增强模型的泛化能力和防止过度拟合SARS-CoV-2特定数据,研究者从BV-BRC数据库中获取了超过1.1亿条独特的原核生物基因序列。 这些序列跨越多个属,代表了广泛的基因多样性。
-
数据选择与整理:通过查询BV-BRC数据库,研究者找到了10,206个独特的PGfams(跨属蛋白质家族),每个家族包含至少30,000个独特的成员。针对每个PGfam,收集了高质量、非冗余的基因和蛋白质序列,这些序列在长度上与该PGfam平均长度相差不超过一个标准差。
-
目的与应用:这些基因级的原核生物数据被用来预先训练更为通用的模型,作为构建SARS-CoV-2特异性GenSLMs的基础。通过这种方式,模型能够在学习SARS-CoV-2特异性进化特性的同时,受益于原核生物基因数据带来的广泛生物信息学背景,从而增强模型的泛化能力。
三、训练结果
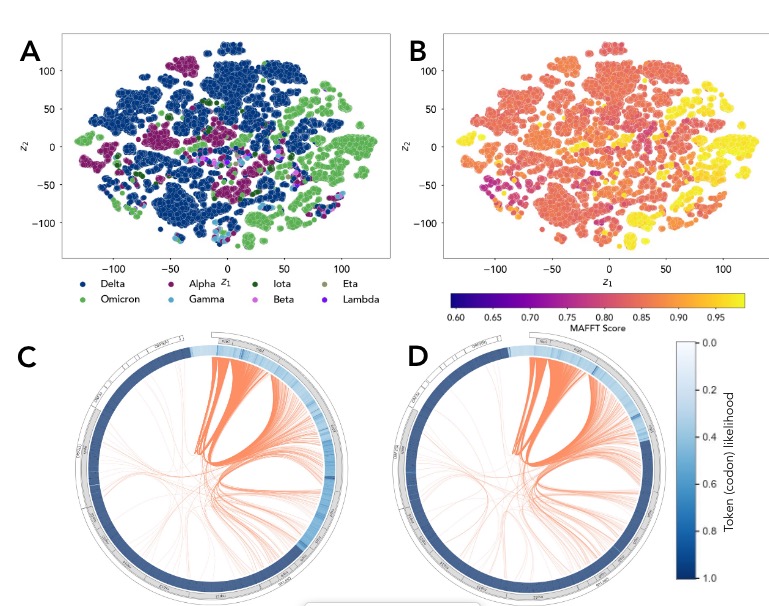
Fig2
模型能够有效区分不同SARSCoV-2到不同变体
作者使用t-SNE将学习到的隐藏空间投射到低维流形上,在Fig2中,居然有效地区分了SARSCoV-2变体,如图2所示。有趣的事,GenSLM-25M模型仅在SARS-CoV-2数据的第一年(由~ 85,000个SARS-CoV-2基因组序列组成)进行了专门训练,并没有机会看到任何其他菌株。 说明了它学习之后的泛化能力。 在硬件上,通过Cerebras CS-2的加持,在独立模式下和作为一个相互连接的集群,并获得了在不到一天的时间内收敛的genslm。
模型生成新序列
GenSLM模型的后续应用在于它能够生成新的SARS-CoV-2序列,甚至预测尚未见过的voc。 这里文章结合了结合了束搜索策略与奖励函数来克服基于序列生成策略中难以采样具有特定属性序列的挑战。
首先,给定一个条件序列模型最可能的序列可以表示为
其中c是来自先前推理的上下文信息。然而,直接计算这个概率通常不可行,因为其复杂度为,其中T是最大序列长度,词汇表大小为64。
在这里,作者使用了条件序列模型生成概率来量化这个问题:作者采用贪心采样(greedy sampling)等启发式方法,即迭代生成序列,每次选择下一个tokens时最大化,则其复杂度降为.
在这个贪心采样中进行束搜索,其中k表示同时探索的“束”(beam)数量,复杂度为 O(kT)。具体操作如下:
-
根据最高概率(或从多项分布中抽样)抽取k个样本,将其加入可能的候选集。对于时间步 t,从所有tokens中选择 k 个得分最高的tokens 根据以下分数函数从中选择最高分的束并输出:
随后,在后面增加一个情景奖励函数,来修改波束搜索的评分函数
在每一步的波束搜索中,选择得分最高的波束
该公式描述了条件序列模型在给定初始上下文c的情况下,生成完整序列x的概率。在最大似然框架下,最大化的概率对应着模型认为最有可能出现的序列。这种评分修改有效地改变了基于最大化奖励函数的令牌被抽样的可能性。实现了对与固定序列相类似的序列进行抽样
通过Diffusion-based来捕获远程依赖关系
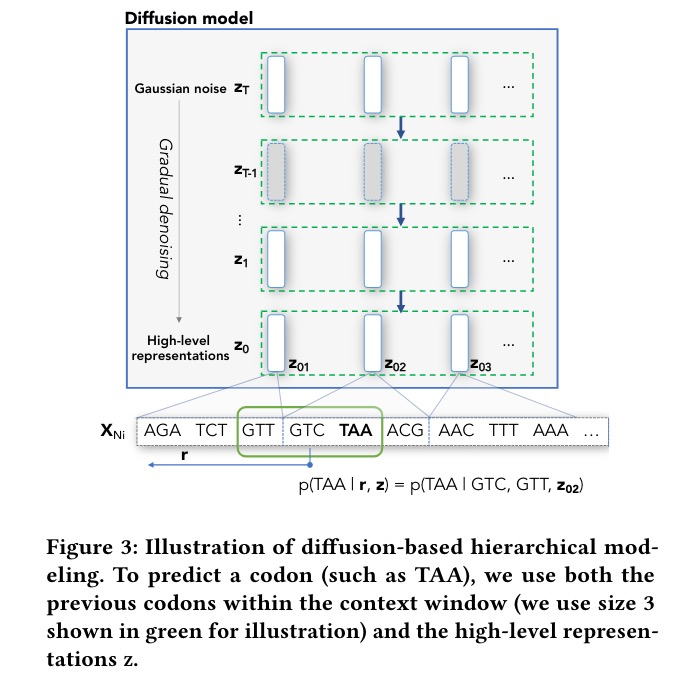
分句与编码: 首先,将完整的基因组序列均匀地划分为多个子序列(每个子序列包含512个密码子),并在相邻子序列之间插入特殊分隔符号。每个这样的子序列被视为一个“句子”。约20个句子对应约17个ORFs和非编码区域.
学习高层表示: 构建一个新的编码器,用于将这些“句子”嵌入到一个潜在空间中。训练过程类似于SpanBERT中的遮蔽语言建模目标,即通过条件上下文预测中间缺失的“句子”,同时使用随机抽取的其他“句子”作为负样本(distractors)。
扩散模型建模高层动态: 在获得每个基因组的“句子”嵌入序列后, 扩散模型通过在高斯噪声之上应用一系列去噪操作来参数化高层表示的分布,采用去噪得分匹配作为训练目标,即逐渐向目标表示添加噪声,然后让模型学习在每一阶段去除噪声。
基于高层的LM微调: 解码器在预测当前密码子时,不仅考虑其局部上下文窗口内的前序密码子,还利用对应的“句子”嵌入。具体操作是将GenSLMs(全基因组规模语言模型)作为解码器进行微调,使其在生成基因组序列时依赖于前面步骤中学习到的高层表示。
作者通过一个生成数据集与真实数据集的比较来反映了模型的能力。ORFs数量的统计分布情况: 一是实际观测到的SARS-CoV-2基因组数据(即真实数据),二是由基于扩散的层次语言模型(diffusion-based hierarchical LM)生成的1,000个SARS-CoV-2基因组序列样本。作者通过覆盖在系统发育树上的模型生成的序列(浅蓝色)表明这些序列与观察到的菌株相似。
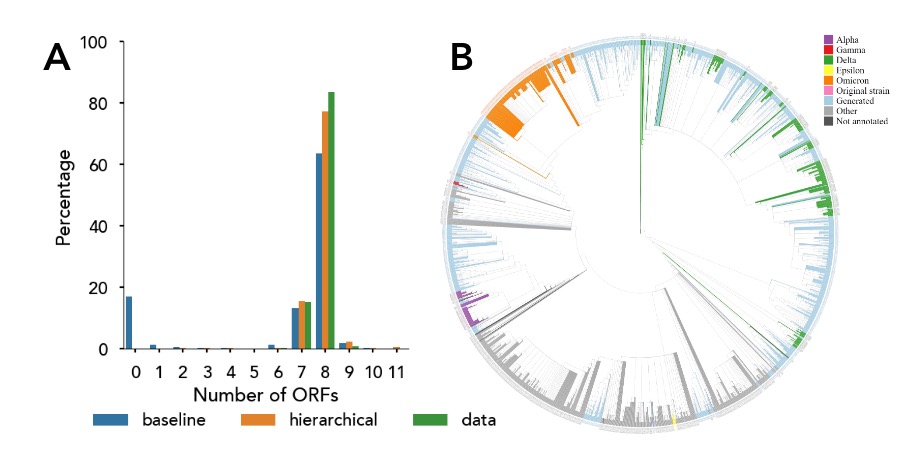
Fig4 基于扩散的层次模型在生成SARS-CoV-2基因组时成功捕捉到了真实的高级动态
模型总结
作者描述了模型的概念性的工作流程概述,展示了如何使用GenSLMs(基因组规模语言模型)和奖励引导的束搜索(reward-guided beam search)方法来生成具有特定属性的SARS-CoV-2序列。

-
Thinker组件:负责在两个关键应用程序之间(即序列生成器和贝叶斯优化器)协调数据流动,确保生成的序列朝着目标属性进行优化。
-
序列生成器:基于GenSLM模型,根据给定的上下文信息和内部状态生成新的SARS-CoV-2序列。生成过程中,模型会考虑先前生成的令牌(tokens)和上下文知识,以生成连贯且可能具有特定属性的序列。
-
奖励引导的束搜索:在生成序列时,引入一个奖励函数来评估生成序列相对于目标属性的符合程度。使用一个混合常数μ(mixing constant)来平衡奖励函数与基于模型概率的生成偏好。μ决定了在选择下一个令牌时,奖励分数对生成决策的影响力。
-
贝叶斯优化器:负责调整奖励混合超参数μ,以使序列生成器对目标属性产生偏倚。通过监控生成序列的性能,更新高斯过程代理模型,并据此建议新的μ值,驱动属性优化过程。
-
工作流程交互:启动时,多个GenSLM生成器进行μ参数空间((0, 1))的初始网格搜索,生成的序列用于更新高斯过程模型。
-
基础设施: 1.资源分配:每个生成任务使用单个A100 GPU执行GenSLM模型实例,而优化任务则使用单节点上的CPU资源。 2.Colmena扩展:通过实现Colmena工作流工具包中的Application抽象,为每个任务提供进程间通信和热启动功能,确保Thinker能够在登录节点上高效协调多实例生成器与优化器之间的任务提交和通信,实现属性优化的稳健执行。
模型评估
此外,本文还基于一些超级计算平台评估了模型的计算性能,非我等平民所能做得起的,所以实验内容就放一放就行
计算平台: 评估了两种领导级GPU超级计算机(Polaris和Selene)以及Cerebras CS-2 Wafer-Scale Cluster的性能,具有强大的计算能力和先进的互连技术。



模型扩展性: 展示了GenSLM模型在增加GPU数量时的弱线性缩放能力,表明模型训练可以有效利用增加的计算资源。
模型收敛速度: 报告了不同规模模型达到收敛所需的时间,以及使用不同优化技术(如ZeRO-3)对收敛速度的影响。
计算负载: 通过分析不同模型规模和序列长度下,模型的计算负载(FLOPs)以及计算与通信开销的比例,揭示了模型训练过程中的性能瓶颈。
内存占用: 考察了模型在训练过程中内存使用情况,包括激活、梯度和参数的存储需求,以及对显存容量的敏感性。
通信效率: 评估了模型在分布式训练环境中的通信效率,包括数据同步、参数更新等操作的开销,以及不同通信库(如NCCL、Gloo)对性能的影响。
四、项目试运行
import torch
import numpy as np
from torch.utils.data import DataLoader
import sys
# 现在可以尝试导入库中的模块
import genslm # 替换为库中实际模块名
from genslm import GenSLM, SequenceDataset
# Load model
model = GenSLM("genslm_25M_patric", model_cache_dir="content/")
model.eval()
# Select GPU device if it is available, else use CPU
device = "cuda" if torch.cuda.is_available() else "cpu"
model.to(device)
# Input data is a list of gene sequences
sequences = [
"ATGAAAGTAACCGTTGTTGGAGCAGGTGCAGTTGGTGCAAGTTGCGCAGAATATATTGCA",
"ATTAAAGATTTCGCATCTGAAGTTGTTTTGTTAGACATTAAAGAAGGTTATGCCGAAGGT",
]
dataset = SequenceDataset(sequences, model.seq_length, model.tokenizer)
dataloader = DataLoader(dataset)
# Compute averaged-embeddings for each input sequence
embeddings = []
with torch.no_grad():
for batch in dataloader:
outputs = model(
batch["input_ids"].to(device),
batch["attention_mask"].to(device),
output_hidden_states=True,
)
# outputs.hidden_states shape: (layers, batch_size, sequence_length, hidden_size)
# Use the embeddings of the last layer
emb = outputs.hidden_states[-1].detach().cpu().numpy()
# Compute average over sequence length
emb = np.mean(emb, axis=1)
embeddings.append(emb)
# Concatenate embeddings into an array of shape (num_sequences, hidden_size)
embeddings = np.concatenate(embeddings)
embeddings.shape
print(embeddings)
print(embeddings.shape)
基于序列进行embedding
Tokenizing...: 100%|██████████| 2/2 [00:00<00:00, 737.65it/s]
[[-1.5554018 0.5398484 -0.3293697 ... 0.5514648 0.84118253
1.0735412 ]
[-1.3410243 0.05953678 -0.5102916 ... 1.2094046 0.01060436
1.0515021 ]]
(2, 512)
输入启动子,进行模型生成
from genslm import GenSLM
# Load model
model = GenSLM("genslm_25M_patric", model_cache_dir="/content/gdrive/MyDrive")
model.eval()
# Select GPU device if it is available, else use CPU
device = "cuda" if torch.cuda.is_available() else "cpu"
model.to(device)
# Prompt the language model with a start codon
prompt = model.tokenizer.encode("ATG", return_tensors="pt").to(device)
tokens = model.model.generate(
prompt,
max_length=10, # Increase this to generate longer sequences
min_length=10,
do_sample=True,
top_k=50,
top_p=0.95,
num_return_sequences=2, # Change the number of sequences to generate
remove_invalid_values=True,
use_cache=True,
pad_token_id=model.tokenizer.encode("[PAD]")[0],
temperature=1.0,
)
sequences = model.tokenizer.batch_decode(tokens, skip_special_tokens=True)
for sequence in sequences:
print(sequence)
>>> ATG GTT ATT TCA TCT GAT TTA CCA ACT
>>> ATG TTC ATT CTT CCG GCA CTT ATC GAA














 1640
1640











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









