







CompareM介绍
CompareM 是一个支持执行大规模比较基因组分析的软件工具包。它为一组基因组(例如,氨基酸同一性)和单个基因组(例如,密码子使用情况)提供统计数据。为了能够处理数千个基因组,计算密集型任务提供了并行化实现。常见的工作流程以单一方法提供,以便用户容易采用,并为有经验的用户提供了更细粒度的接口,以利用特定功能。CompareM 是开源的,根据 GNU 通用公共许可证(版本 3)发布。
功能
比较基因组统计:
基因组间的平均氨基酸一致性(AAI)
通过计算查询基因组与参考数据库之间的AAI进行分类学分类
基因组使用模式:
密码子使用
氨基酸使用
对 k ≤ 8 的 kmer 使用(例如,四核苷酸)
终止密码子使用
其他:
用于识别水平基因转移(LGT)的二核苷酸和密码子使用模式
使用不相似性矩阵、层次聚类树和热图进行数据探索
安装
compareM依赖的python环境:3.6
**注意:**本人第一次安装是在python=3.11环境,安装成功运行时,提示错误:RecursionError: maximum recursion depth exceeded while calling a Python object。
conda create -n comparem python=3.6
conda activate comparem
conda install comparem
comparem -h
使用
1. 计算氨基酸使用
comparem aa_usage -h
usage: comparem aa_usage [-h] [–counts] [-x FILE_EXT] [-c CPUS] [–silent]
protein_gene_files output_file
Calculate amino acid usage within each genome.
positional arguments:
protein_gene_files input files with genes in amino acid space
output_file output file indicating amino acid usage for each
genome
optional arguments:
-h, --help show this help message and exit
–counts output raw counts instead of frequencies
-x, --file_ext FILE_EXT
extension of files to process (default: faa)
-c, --cpus CPUS number of CPUs to use (default: 1)
–silent suppress output
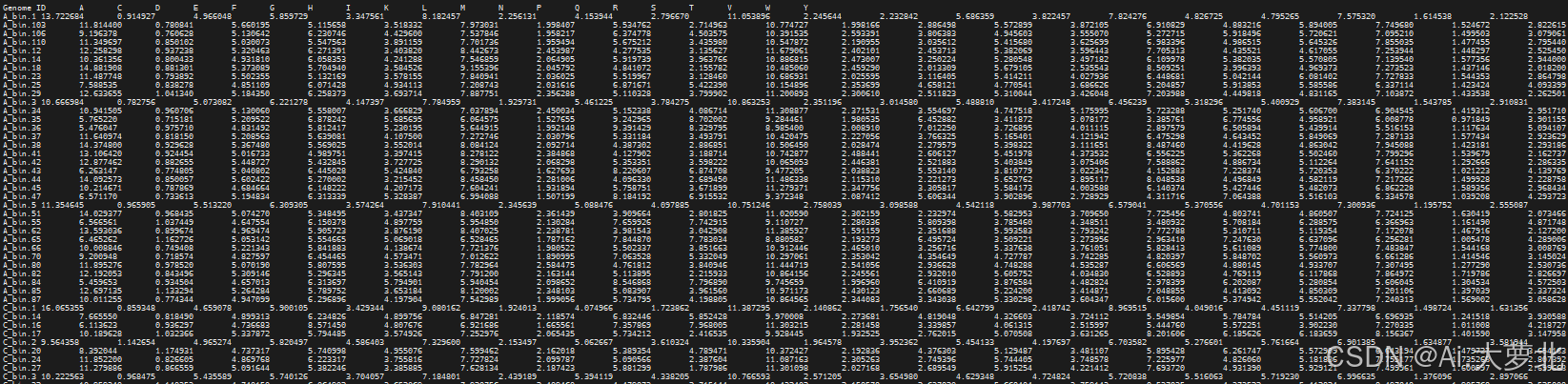
$ comparem aa_usage 9phylum_244.bins comparem.out
[2025-01-10 09:43:17] INFO: CompareM v0.1.2
[2025-01-10 09:43:17] INFO: comparem aa_usage 9phylum_244.bins comparem.out
[2025-01-10 09:43:18] INFO: Calculating amino acid usage for each genome:
Finished processing 244 of 244 (100.00%) genomes.
[2025-01-10 09:44:34] INFO: Amino acid usage written to: comparem.out
输入文件protein_gene_files(9phylum_244.bins),是需要计算氨基酸usage的基因组地址列表
输出文件,将所有基因组的结果重定向到一个文件。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









