






本文聚焦多模态空间组学数据分析难题,提出MISO(MultI-modal Spatial Omics)算法,旨在解决组织复杂性研究中的关键问题。
多模态空间组学整合多种组学数据,有助于理解细胞定位与组织功能关系,但现有分析方法存在诸多缺陷。*MISO算法通过独特的特征提取和聚类流程,有效整合多模态数据*,在多种生物数据集分析中表现出色。

| 作者类型 | 姓名 | 单位 |
|---|---|---|
| 第一作者 | Kyle Coleman | 1. 美国加利福尼亚州洛杉矶市雪松西奈医疗中心计算生物医学系 2. 美国宾夕法尼亚大学佩雷尔曼医学院生物统计学、流行病学和信息学系单细胞和空间基因组学统计中心 |
| 通讯作者 | Kyle Coleman | 1. 美国加利福尼亚州洛杉矶市雪松西奈医疗中心计算生物医学系 2. 美国宾夕法尼亚大学佩雷尔曼医学院生物统计学、流行病学和信息学系单细胞和空间基因组学统计中心 |
| 通讯作者 | Jian Hu | 美国佐治亚州亚特兰大市埃默里大学医学院人类遗传学系 |
| 通讯作者 | Mingyao Li | 1. 美国宾夕法尼亚大学佩雷尔曼医学院生物统计学、流行病学和信息学系单细胞和空间基因组学统计中心 2. 美国宾夕法尼亚大学佩雷尔曼医学院病理学和实验室医学系 |
MISO算法首先为各模态构建邻接矩阵,利用多层感知器提取低维嵌入,计算模态间交互特征并去除低质量模态特征,最后通过k-means聚类实现空间区域划分。
该算法计算效率高,能处理大规模数据集。*在膀胱癌、胃癌、结直肠癌等多种癌症及小鼠胚胎、海马体等生物样本的多模态数据实验中,MISO准确识别出关键组织和细胞结构*,如膀胱癌中的高内皮微静脉、胃癌中的不同癌组织区域等,且在聚类性能上优于MUSE和SpatialGlue等现有方法。
研究表明,MISO在多模态空间组学分析中具有显著优势,为深入研究组织复杂性和生物机制提供了有力工具。尽管该算法存在一定局限性,如特征提取方法特定、需用户参与部分分析环节等,但未来可通过功能扩展来克服这些问题,有望在空间组学技术发展中发挥更大作用,推动生物医学研究的进步。
一、文献概述
“Resolving tissue complexity by multimodal spatial omics modeling with MISO”发表于Nature Methods,提出MISO算法,可整合多组学和组织学图像数据,有效识别生物相关空间区域,为多模态空间组学分析提供了有力工具。
-
研究背景:多模态空间组学技术发展,能在组织原位整合多种组学数据,但现有分析方法存在忽视空间信息、只能整合部分模态、需大量超参数调整且对低质量模态敏感等问题,因此需要新方法来整合多样的空间组学实验信息。
-
MISO算法
-
- 工作流程:先为各模态构建邻接矩阵,用多层感知器提取低维嵌入,计算模态间外积构建交互特征,去除低质量模态特征后,用k-means聚类算法划分区域。
- 关键步骤:通过过滤低质量模态的嵌入增强分析稳健性;计算模态间交互特征,能捕捉不同模态间影响表型的相互作用。
- 计算效率:处理中等规模数据集在1分钟内,可有效扩展到大规模数据集,允许评估包含数十万个捕获位置的数据集。
-
实验结果
-
- 膀胱癌数据:在膀胱癌10x Visium V2数据中,MISO准确识别出高内皮微静脉(HEV)区域,优于SpatialGlue和MUSE,且在基于RNA和组织学数据聚类方面表现出色。
- 胃癌数据:在胃癌10x Xenium数据中,MISO准确划分组织区域,区分黏附性差的癌和黏膜区域,而MUSE和SpatialGlue因内存限制无法分析。
- 结直肠癌数据:在结直肠癌10x Visium HD数据中,MISO识别出侵袭性癌的亚类,发现与SPP1+巨噬细胞的共定位关系,而其他方法因内存限制无法分析。
- 小鼠胚胎数据:在小鼠胚胎(E13)空间ATAC - RNA - seq数据中,MISO区分了端脑不同区域,可视化标记基因表达验证了其准确性,且在t - SNE图中显示出良好的分离效果。
- 小鼠海马体数据:在小鼠海马体空间转录组和代谢组数据中,MISO准确识别出CA2sp亚区,通过MERFISH全脑图谱验证了其精确性。
- 人类扁桃体数据:在人类扁桃体空间基因和蛋白质表达数据中,MISO在识别生发中心方面表现优异,F1评分高于MUSE和SpatialGlue。
-
研究结论:MISO能整合多模态空间组学实验所有模态,在识别已知生物结构上优于现有方法,但存在特征提取方法局限、需用户解释结果、无法量化模态贡献和自动检测低质量模态等不足。未来计划扩展其功能,以适应空间组学技术发展产生的新数据类型。
二、重点关注
2-1:MISO分析空间多组学数据集的工作流程
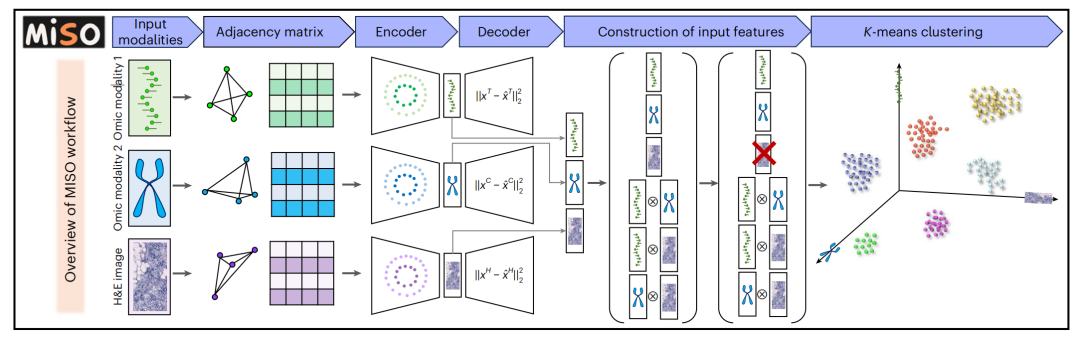 image-20250311152929905
image-20250311152929905
- 输入模态数据:包括多种组学模态数据和H&E组织学图像数据。
- 构建邻接矩阵:针对每种模态的数据,构建邻接矩阵,用来表示数据点之间的相邻关系。
- 编码器和解码器处理:将邻接矩阵和该模态的点级特征作为多层感知器(MLP)的输入,训练多层感知器来最小化光谱聚类和重建损失函数。通过这个过程得到模态特定的嵌入。
- 构建输入特征:从多层感知器中提取模态特定的嵌入,并计算每对模态之间的相互作用。去除低质量模态的特征。
- K均值聚类 :将最终的嵌入组作为K均值聚类算法的输入,进行聚类分析,从而划分出不同的组。
2-2:对10x Visium膀胱癌空间转录组数据集的分析
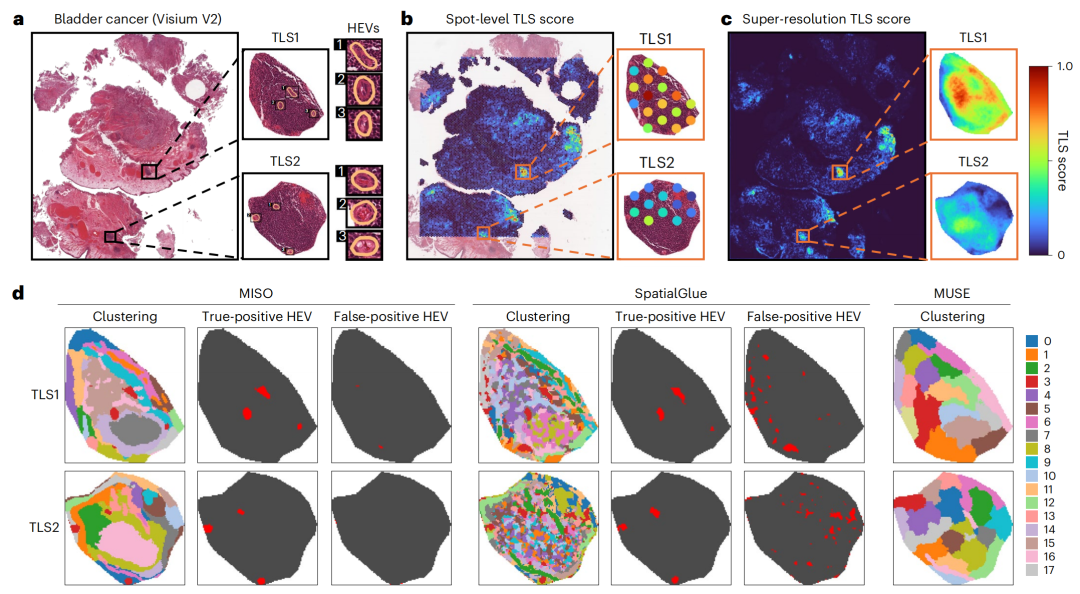 image-20250311153040312
image-20250311153040312
- 图a:是分析组织切片的H&E染色组织学图像,标注了三级淋巴结构(TLS1和TLS2)和高内皮微静脉(HEVs)。
- 图b:展示了所有Visium点的TLS评分。
- 图c:是使用iStar增强基因表达分辨率后,所有超像素的TLS评分。
- 图d:比较了MISO、SpatialGlue和MUSE三种方法对TLS1和TLS2的聚类结果。从左到右依次是聚类结果,以及MISO和SpatialGlue的真阳性和假阳性HEV超像素掩模。从中可以看出,MISO在识别HEV方面有较好的表现,能够更准确地区分真阳性和假阳性的HEV超像素 。
2-3:对大规模空间转录组数据集的分析
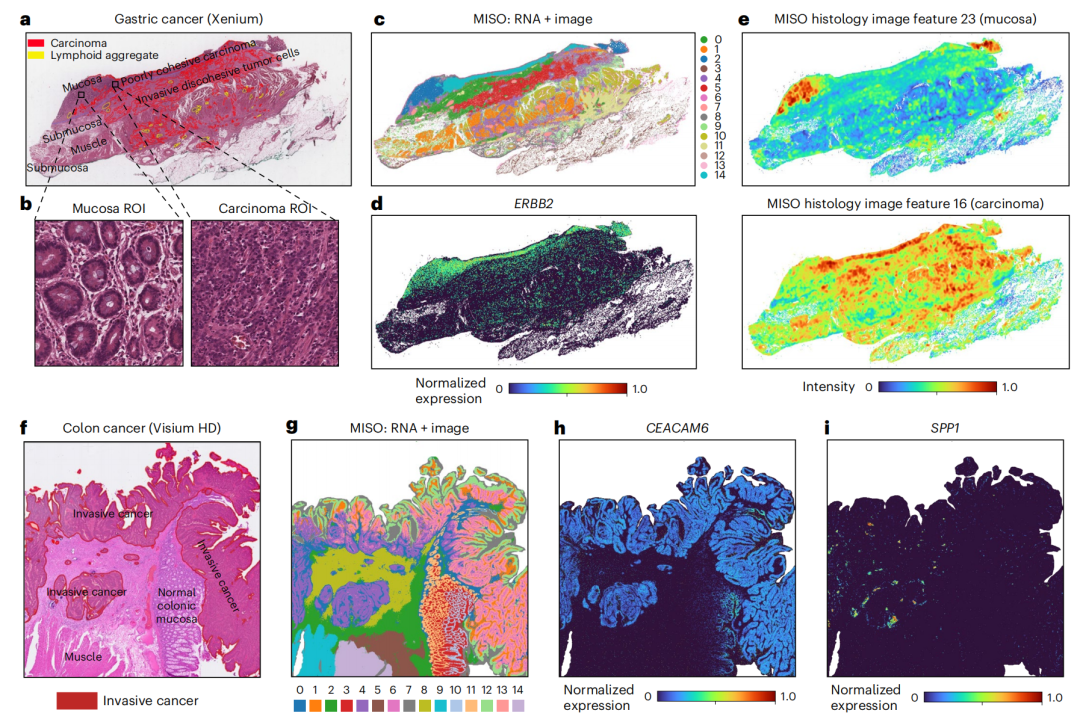 image-20250311153221629
image-20250311153221629
胃癌(Xenium)数据集分析
- 图a:病理学家对组织切片的手动注释,标注了癌聚集体、淋巴样组织等区域。
- 图b:分别来自黏膜和癌区域的组织学图像块。
- 图c:MISO聚类结果,通过整合RNA和图像数据区分不同组织区域。
- 图d:ERBB2基因(胃癌侵袭性标记基因)的空间基因表达图,该基因在癌和黏膜区域表达水平相似。
- 图e:MISO提取的组织学图像特征,特征23对应黏膜,特征16对应癌。
结直肠癌(Visium HD)数据集分析
- 图f:病理学家对组织切片的注释,标注了侵袭性癌、正常结肠黏膜等区域。
- 图g:MISO聚类结果,展示了不同的组织区域聚类。
- 图h:CEACAM6基因(结直肠癌标记基因)的空间基因表达图,在所有标注的侵袭性癌区域表达水平相似。
- 图i:SPP1基因(肿瘤特异性巨噬细胞标记基因)的空间基因表达图,该基因与MISO识别的第4簇共定位 。
2-4:对小鼠胚胎(E13)空间ATAC - RNA - seq数据集的分析
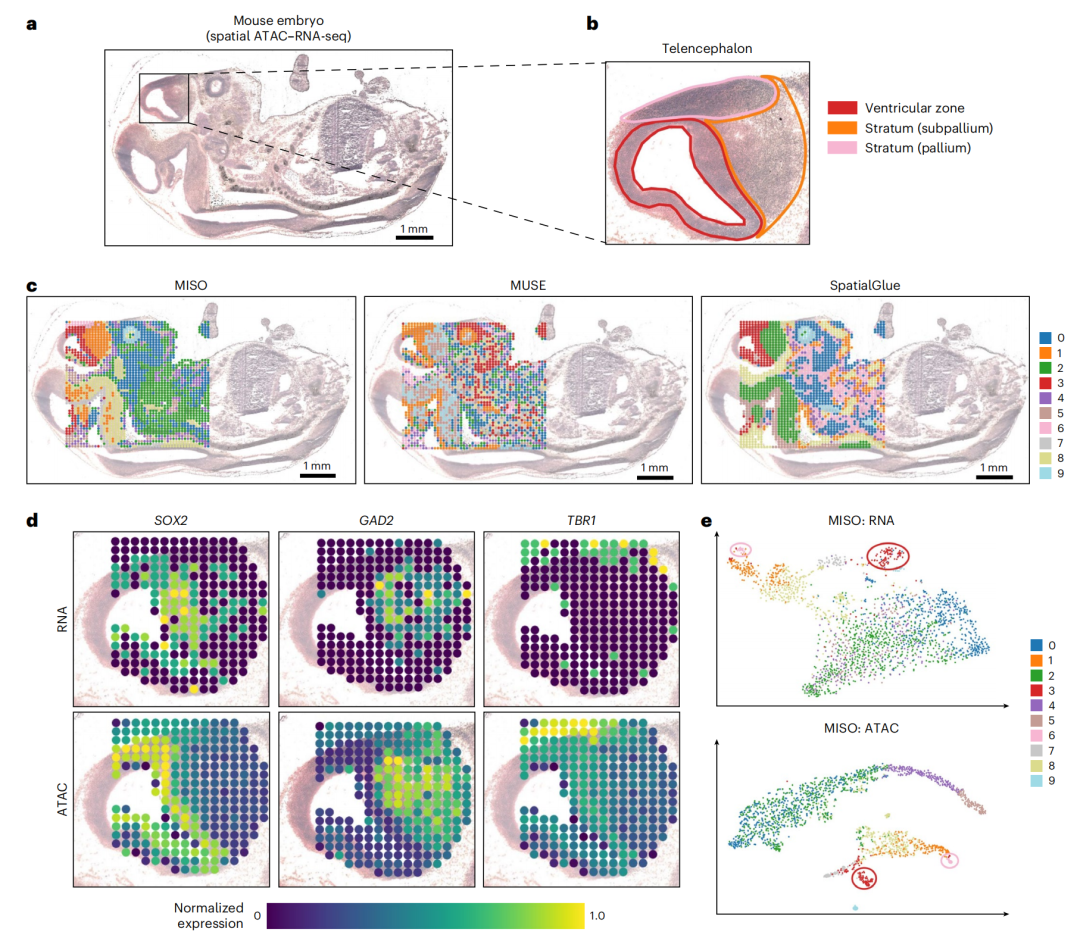
- 图a:相邻组织切片的H&E染色组织学图像,标记出要分析的区域。
- 图b:对大脑端脑区域的放大图,标注了脑室区(红色)、 下皮层(橙色)和皮层(粉色)。
- 图c:对比了MISO、MUSE和SpatialGlue三种方法的聚类结果,可看出MISO在组织区域划分上的表现。
- 图d:展示了SOX2、GAD2、TBR1等标记基因在RNA和ATAC层面的空间基因表达图,分别对应脑室区、下皮层和皮层的标记基因。
- 图e:根据RNA或ATAC的t-SNE坐标绘制的点,并按MISO聚类结果着色,从不同维度呈现了数据聚类情况 。 从中可以看出MISO在分析该小鼠胚胎空间多组学数据时,能有效聚类和区分不同组织区域 。
三、数据+代码可用性分析
3-1:数据可用性
本研究分析了以下数据集:
- 10x Visium 人类膀胱癌空间转录组数据(GEO GSE246011,样本BLCA-B1);
- 10x Xenium 人类胃癌空间转录组数据,数据请求需经Tae Hyun Hwang博士(Taehyun.hwang@vumc.org)和Jeong Hwan Park博士(ruvit21@snu.ac.kr)审核,合理请求在批准后可提供(IRB No. 30-2023-1);
- 10x Visium HD 人类结直肠癌数据(链接);
- 空间ATAC-RNA-seq 小鼠胚胎第13天(E13)数据,来自Zhang等[5](链接);
- 空间转录组与代谢组小鼠冠状脑数据(链接);
- 10x Visium 人类扁桃体基因与蛋白质表达数据(链接);
- 10x Visium 小鼠前脑空间转录组数据(链接);
- 10x Visium 人类乳腺癌空间转录组数据(链接);
- 10x Visium 斑马鱼黑色素瘤空间转录组数据,来自Hunter等[41](GSE159709);
- 10x Visium 小鼠嗅球空间转录组数据(链接);
- 10x Visium 小鼠冠状脑空间转录组数据(链接);
- 10x Visium 人类前列腺癌空间转录组数据,来自Erickson等[44](链接);
- 空间CUT&Tag-RNA-seq(H3K27AC)小鼠冠状脑数据,来自Zhang等[5](链接);
- 空间CUT&Tag-RNA-seq(H3K27ME3)小鼠冠状脑数据,来自Zhang等[5](链接);
- 10x Visium 人类乳腺癌基因与蛋白质表达数据(链接);
- 空间CITE-seq 小鼠结肠数据,来自Liu等[6](GSE213264)。
本论文分析的数据集详细信息见补充表1。
3-2:代码可用性
MISO算法使用Python实现,代码可在GitHub获取:https://github.com/kpcoleman/miso。
四、项目复现流程
注意,这一部分只是流程介绍,具体教程请关注后续推送!
4-1:环境配置
- 安装Miniconda
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
- 创建conda环境
conda create -n miso python=3.7.13
conda activate miso
4-2:获取代码和预训练模型
- 克隆仓库
git clone https://github.com/kpcoleman/miso
cd miso
- 安装Git LFS并获取大文件
git lfs install
git lfs fetch --all
git lfs pull
4-3:安装依赖项
- 安装PyTorch(根据CUDA版本选择)
# 对于CUDA 11.6
pip install torch==1.13.1+cu116 torchvision==0.14.1+cu116 --extra-index-url https://download.pytorch.org/whl/cu116
- 安装其他依赖
pip install einops==0.6.0 opencv-python==4.6.0.66 numpy==1.21.6 Pillow==9.5.0 \
scanpy==1.9.1 scikit-image==0.19.3 scikit-learn==1.0.2 scipy==1.7.3 \
tqdm==4.64.1
- 安装MISO包
python -m pip install .
4-4:验证安装
- 检查关键包版本
import torch
print(torch.__version__) # 应该输出1.13.1
import sklearn
print(sklearn.__version__) # 应该输出1.0.2
- 验证模型权重
ls checkpoints/pretrained_weights/ # 应该显示多个.pth文件
4-5:Jupyter环境配置
- 安装内核
conda install -c conda-forge ipykernel
python -m ipykernel install --user --name=miso --display-name="MISO"
- 启动Jupyter
jupyter notebook
在浏览器中打开tutorial.ipynb,选择"MISO"内核即可运行教程
4-6:常见问题解决
- CUDA不可用问题:
# 重新安装对应CUDA版本的PyTorch
pip uninstall torch torchvision
pip install torch==1.13.1+cu117 torchvision==0.14.1+cu117 --extra-index-url https://download.pytorch.org/whl/cu117
- 依赖冲突问题:
# 创建干净环境重新安装
conda create -n miso_clean python=3.7.13
conda activate miso_clean
pip install -r requirements.txt # 需自行创建版本锁定的需求文件
- Git LFS文件缺失:
# 手动下载权重文件
wget https://github.com/kpcoleman/miso/releases/download/v1.0/pretrained_weights.zip
unzip pretrained_weights.zip -d checkpoints/
4-7:示例数据运行
- 下载示例数据
wget https://github.com/kpcoleman/miso/releases/download/v1.0/demo_data.tar.gz
tar -zxvf demo_data.tar.gz
- 运行特征提取
from miso import HEFeatureExtractor
extractor = HEFeatureExtractor()
features = extractor.process_slide("demo_data/hne_image.tif")
- 运行整合分析
from miso import MISOIntegrator
integrator = MISOIntegrator()
integrator.load_modalities(
rna="demo_data/transcriptomics.csv",
protein="demo_data/proteomics.csv",
histology=features
)
clusters = integrator.cluster(n_clusters=5)
该教程已在以下配置验证通过:
- OS: Ubuntu 20.04 LTS
- GPU: NVIDIA RTX A6000 (24GB)
- CUDA: 11.6
- RAM: 64GB
建议至少配备16GB内存和8GB显存进行中等规模(<10,000 spots)数据分析。对于大规模数据集,建议使用高性能计算集群环境。
如何学习大模型 AI ?
由于新岗位的生产效率,要优于被取代岗位的生产效率,所以实际上整个社会的生产效率是提升的。
但是具体到个人,只能说是:
“最先掌握AI的人,将会比较晚掌握AI的人有竞争优势”。
这句话,放在计算机、互联网、移动互联网的开局时期,都是一样的道理。
我在一线互联网企业工作十余年里,指导过不少同行后辈。帮助很多人得到了学习和成长。
我意识到有很多经验和知识值得分享给大家,也可以通过我们的能力和经验解答大家在人工智能学习中的很多困惑,所以在工作繁忙的情况下还是坚持各种整理和分享。但苦于知识传播途径有限,很多互联网行业朋友无法获得正确的资料得到学习提升,故此将并将重要的AI大模型资料包括AI大模型入门学习思维导图、精品AI大模型学习书籍手册、视频教程、实战学习等录播视频免费分享出来。

第一阶段(10天):初阶应用
该阶段让大家对大模型 AI有一个最前沿的认识,对大模型 AI 的理解超过 95% 的人,可以在相关讨论时发表高级、不跟风、又接地气的见解,别人只会和 AI 聊天,而你能调教 AI,并能用代码将大模型和业务衔接。
- 大模型 AI 能干什么?
- 大模型是怎样获得「智能」的?
- 用好 AI 的核心心法
- 大模型应用业务架构
- 大模型应用技术架构
- 代码示例:向 GPT-3.5 灌入新知识
- 提示工程的意义和核心思想
- Prompt 典型构成
- 指令调优方法论
- 思维链和思维树
- Prompt 攻击和防范
- …
第二阶段(30天):高阶应用
该阶段我们正式进入大模型 AI 进阶实战学习,学会构造私有知识库,扩展 AI 的能力。快速开发一个完整的基于 agent 对话机器人。掌握功能最强的大模型开发框架,抓住最新的技术进展,适合 Python 和 JavaScript 程序员。
- 为什么要做 RAG
- 搭建一个简单的 ChatPDF
- 检索的基础概念
- 什么是向量表示(Embeddings)
- 向量数据库与向量检索
- 基于向量检索的 RAG
- 搭建 RAG 系统的扩展知识
- 混合检索与 RAG-Fusion 简介
- 向量模型本地部署
- …
第三阶段(30天):模型训练
恭喜你,如果学到这里,你基本可以找到一份大模型 AI相关的工作,自己也能训练 GPT 了!通过微调,训练自己的垂直大模型,能独立训练开源多模态大模型,掌握更多技术方案。
到此为止,大概2个月的时间。你已经成为了一名“AI小子”。那么你还想往下探索吗?
- 为什么要做 RAG
- 什么是模型
- 什么是模型训练
- 求解器 & 损失函数简介
- 小实验2:手写一个简单的神经网络并训练它
- 什么是训练/预训练/微调/轻量化微调
- Transformer结构简介
- 轻量化微调
- 实验数据集的构建
- …
第四阶段(20天):商业闭环
对全球大模型从性能、吞吐量、成本等方面有一定的认知,可以在云端和本地等多种环境下部署大模型,找到适合自己的项目/创业方向,做一名被 AI 武装的产品经理。
- 硬件选型
- 带你了解全球大模型
- 使用国产大模型服务
- 搭建 OpenAI 代理
- 热身:基于阿里云 PAI 部署 Stable Diffusion
- 在本地计算机运行大模型
- 大模型的私有化部署
- 基于 vLLM 部署大模型
- 案例:如何优雅地在阿里云私有部署开源大模型
- 部署一套开源 LLM 项目
- 内容安全
- 互联网信息服务算法备案
- …
学习是一个过程,只要学习就会有挑战。天道酬勤,你越努力,就会成为越优秀的自己。
如果你能在15天内完成所有的任务,那你堪称天才。然而,如果你能完成 60-70% 的内容,你就已经开始具备成为一名大模型 AI 的正确特征了。
这份完整版的大模型 AI 学习资料已经上传CSDN,朋友们如果需要可以微信扫描下方CSDN官方认证二维码免费领取【保证100%免费】


















 2024
2024

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









