 肽类药物治疗PPI相关疾病的研究进展
肽类药物治疗PPI相关疾病的研究进展




针对蛋白质-蛋白质相互作用(PPI)的治疗方法最近被公认为治疗多种疾病的新兴疗法。迄今为止,已发现超过50万种PPI失调与病理事件有关。这些过程的动态特性以及大蛋白质表面的参与,使得科学界对它们作为有前景的治疗靶点持谨慎态度。最近,由于药物发现提供了广泛的结构多样化的序列,从传统的内源性肽到具有改善的药理性能的序列,肽类药物重新受到关注。目前,约有70种肽类药物已上市,但还有几种正在临床开发中。在这篇综述中,我们希望报告这些新型API的最新进展,重点关注临床开发中的分子,这是过去10年药物发现过程的直接结果。这些全面的收集将根据结构特征(天然、类似、异源)和治疗靶点进行分类。关于 PPI 干扰机制的报告也将为新型肽的设计提供有用的信息。
在许多病理状态的发展过程中,蛋白质 - 蛋白质相互作用(PPI)起着信号传递介质的作用,正因如此,它们成为药物发现的理想靶点。
人类相互作用组(即蛋白质相互作用的复杂网络)的大小已被估计约为 65 万个相关接触点,这些接触点调节着所有生物事件,当失调时会导致大量病理疾病。蛋白质相互作用的动态特性以及合作伙伴之间通常需要的大接触区域(约 1000 - 4000Å)在过去阻碍了将其视为有趣的目标。小分子药物确实只能抑制涉及 300 至 1000 Å结合区域的相互作用,而且蛋白质相互作用往往缺乏明确的pockets。通常,蛋白质之间的关联发生在被称为“热点”的疏水区域,这些区域富含能够形成氢键和π相互作用的氨基酸,并且只有少数关键残基在决定结合亲和力和特异性方面起作用。许多热点核心区域与α-螺旋、β-折叠和β-转角等蛋白质二级结构的存在有关。
在过去几年中,针对蛋白质-蛋白质相互作用的基序和肽模拟物已被定制,以模拟这些有序结构。
在这种情况下,肽类化合物被排除在潜在的先导化合物之外,因为它们受到几个问题的影响,即短序列的随机构象、长序列合成和纯化的困难以及对内肽酶的敏感性,这通常决定了它们的半衰期只有几分钟。此外,对蛋白水解降解的敏感性使得口服给药成为一个挑战。可靠的技术的发展使得即使是高氨基酸含量的肽类化合物也能达到药用级标准。此外,通过引入非天然氨基酸、引入亲脂性侧链或环状序列,分子设计可以轻松克服上述限制。因此,在过去二十年里,肽类化合物重新受到关注,迄今为止,已有约70种治疗性肽类化合物获得批准并上市。在多肽段新化学实体(NCE)发展的原因中,GLP-1 类似物利拉鲁肽和司美格鲁肽在治疗 2 型糖尿病和肥胖症方面的商业和治疗上的成功是一个重要因素,2020 年销售额超过 76 亿美元。利拉鲁肽的 13 小时半衰期和司美格鲁肽的 7 天半衰期也是口服可用的,这是这些药物成功的基础。多项市场研究预测,从 2019 年的 290 亿美元销售额到 2025 年的 480 亿美元销售额(不包括胰岛素),多肽段将持续全球增长并取得成功,年增长率为 10%。
本综述重点关注过去三年内临床试验中的肽类药物,这是过去十年中蛋白质-蛋白质相互作用(PPI)知识增加以及合成设计和策略演变的直接应用。根据美国食品药品监督管理局(FDA)的定义,仅讨论长度不超过 40 个氨基酸的治疗性肽类药物(FDA - 美国食品药品监督管理局,2018 年)。我们严格考虑针对 PPI 的治疗性肽类药物,不包括诊断性、放射治疗药物、疫苗以及具有不同化学模态的缀合肽。
在临床阶段鉴定肽并非易事。本次调查中讨论的正在研发中的肽的清单是对来自 Cortellis-Clarivate、临床试验数据库(美国和欧洲)以及公司网站的信息进行综合和批判性评估的结果,目的是真正了解产品的状况。事实上,在药物发现和开发过程中,很少有公司通报将新化学实体(NCE)从研发线中剔除的情况,大多数时候这些分子只是从官方报告和网站中消失。此外,由于产品销售、公司间的联合开发、公司并购或国际非专有名称的定义,同一种分子可能会有几个编码和名称。讨论的分子清单包括处于不同临床阶段的 58 种肽:13 种进入1期,26 种处于2期,15种处于3 期,而 4 种即将获得批准,因为新药申请(NDA)已提交给监管机构(图 1)。与过去相比,一个显著的差异是氨基酸数量的增加,在已公开的结构中(58 种结构中有 47 种),含 20 - 40 个残基的肽的数量多于较短的肽(分别为 26 种和 21 种)。然而,考虑到有 9 种未公开的结构属于糖尿病和肥胖症类别,而这类肽通常长度超过 20 个氨基酸,这一数字应该会更高。
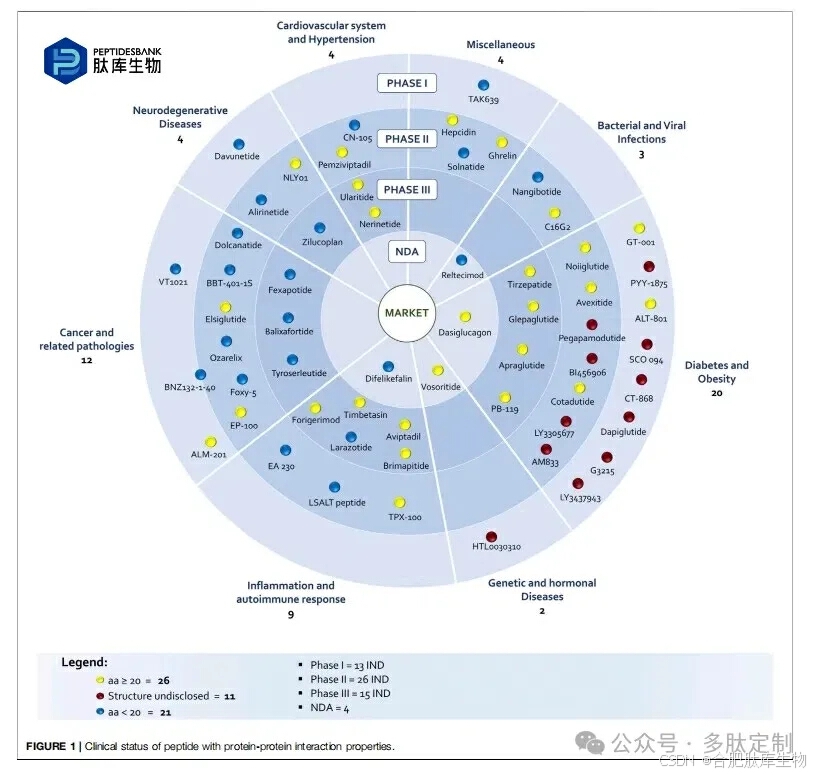
除了通过重组技术生产的两种肽类药物GT001和艾塞那肽外,所有其他披露序列的化合物均为合成生产,通常通过固相合成法(SPPS)进行。在此背景下,全球许多研究小组都在致力于绿色创新技术。至于病理方面,糖尿病、肥胖症、癌症及相关病理是主要的关注领域。
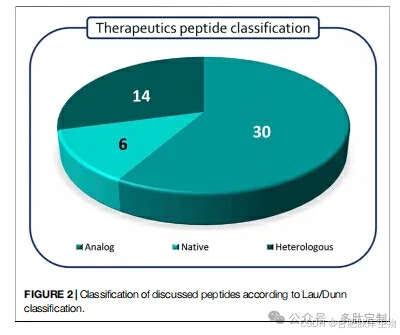
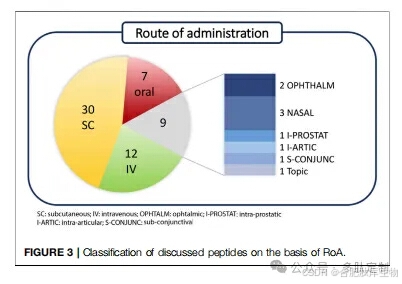
多肽根据劳/邓恩定义进行了分类:天然肽具有与内源性配体相同的序列,而类似物则是为获得更好的药理特征而对天然序列进行修改的产品。异源肽是独立于天然配体发现的,并且基于更经典的药物化学方法(图 2)。值得注意的是,天然肽及其类似物在临床试验中占蛋白质-蛋白质相互作用(PPI)靶向肽的 70%以上,特别是类似物占研发线的 60%。基于目标病理,并且除了临床试验阶段外,还考虑了给药途径(RoA),这是肽类药物代动力学特性的一个体现(图 3)。
癌症及相关病理
蛋白质-蛋白质相互作用(PPI)在肿瘤发展中的作用与蛋白质介导的信号传导过程密切相关,能够激活与肿瘤发生、进展、侵袭和转移相关的多个生物网络。因此,引入对 PPI 的干扰是阻断癌症相关现象的有效策略。观察不同的肿瘤表型,特定的蛋白质在成为致癌现象的一部分时,其相互作用模式会发生变化,这对患者的健康状况产生重要影响。根据肿瘤的位置,可以针对不同的候选蛋白质,通常是跨膜受体或释放激素受体,这些具有诊断和治疗性能,有助于了解癌症进展并提高治疗效果。
关于跨膜受体在癌症发展中的作用,多尔卡纳肽是为治疗炎症性肠病和功能性胃肠疾病以预防结肠癌而设计的。事实上,属于肠上皮的细胞负责通过消化系统控制有毒病原体的排出,以及营养物质、液体和细菌菌群的运输。
并且沃尔德曼(2018 年)指出。为了维持这些过程的体内平衡控制,肠上皮受体鸟苷酸环化酶 C(GUCY2C)及其环核苷酸第二信使环鸟苷酸(cGMP)起着关键作用。这种异源二聚体跨膜受体可以被内源性细胞外肽配体或其他信号因子激活,例如Ca2+和一氧化氮。已知GUCY2C受体的配体是细菌热稳定肠毒素 ST(旅行者腹泻的病因)以及内源性肽类物质尿鸟苷酸和鸟苷酸。Dolcanatide肽被设计为尿鸟苷酸的类似物,由于存在二硫键,对肠道中蛋白酶的标准消化分解具有增强的抗性。该肽的序列与尿鸟苷酸不同,在 N 末端第 3 位用谷氨酸取代天冬氨酸,以提高结合亲和力,并且在 N 末端和 C 末端分别用 D-天冬氨酸和 D-亮氨酸取代 L-天冬氨酸和 L-亮氨酸,据认为这可增强生物稳定性。Dolcanatide肽应该会像尿鸟苷酸和已知的其他内源性配体一样与 GUCY2C 结合,通过 NH2-terminalβ-发夹结构,提供一个小的三股反平行的β-折叠的第三链。
同样,BBT - 401(结构未公开)是一种口服的 Pellino - 1 蛋白 - 蛋白相互作用抑制剂,用于治疗受溃疡性结肠炎影响的患者。这种脂化四肽与 Pellino-1 蛋白结合,干扰其信号级联反应。BBT-401 能有效抑制脂多糖诱导的 Toll 样受体促炎信号通路的激活,并减少促炎细胞因子的表达。对动物模型的研究表明,在结肠炎中给予 BBT-401 显著改善了与该疾病相关的症状和组织病理学参数,而直接结肠给药大大增强了治疗效果。
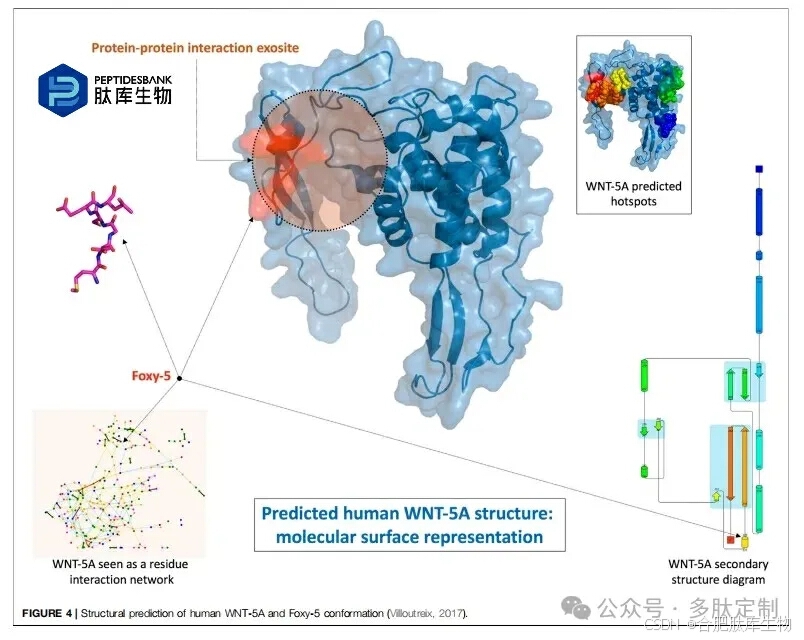
干扰表面受体功能似乎与实体瘤和转移性肿瘤的进展特别相关。Foxy-5是一种甲酰化的WNT5A衍生六氨基酸肽,最近在转移性乳腺癌、结肠癌和前列腺癌患者的1期试验以及早期结肠癌的2期试验中得到应用。其模型蛋白WNT5A是Wnt蛋白家族的一员,在器官发育、组织定向、细胞极性和迁移等生理现象中发挥重要作用。由于其在结肠癌、神经母细胞瘤、乳腺癌和白血病中的肿瘤抑制功能,其失调与各种疾病的进展有关。在这种情况下,Foxy-5被描述为一种模拟WNT5A的肽,因为它通过触发胞质游离钙信号传导和损害上皮癌细胞的迁移和侵袭来发挥作用。通过计算预测,Foxy-5在溶液中可能采取短环和α-螺旋结构,这是蛋白质-蛋白质相互作用中常见的基序。在全长天然蛋白的结构中,这一段暴露于溶剂中,可能参与大分子相互作用。Foxy-5 似乎是外位点的一部分,预计对于 WNT-5A 与受体的相互作用至关重要,并且是二聚位点的一部分(图 4)。
VT1021 是由 Vigeo Therapeutics 公司开发的一种环五肽,其结构仍未公开。它能够有力地诱导血小板反应蛋白 - 1(Tsp - 1)的表达。肿瘤微环境(TME)随后会完全重新编程,以降低其免疫抑制和肿瘤促进倾向,转而促进适应性免疫系统的激活及其抑制肿瘤的倾向。VT-1021 正处于治疗实体瘤的临床试验 1 期,因为在临床前动物模型中,它对卵巢癌、胰腺癌和乳腺癌显示出强大的抗肿瘤活性,肿瘤完全消退,免疫肿瘤微环境也重新编程。
Balixafortide 是另一种强效且选择性的趋化因子受体 CXCR4 拮抗剂的良好范例,CXCR4 属于 G 偶联蛋白受体(GPCR)的跨膜受体家族。这种蛋白质在多种肿瘤类型中大量表达,包括在致癌组织中免疫系统细胞的表达。CXCR4 在转移现象中的作用尤为重要,因为这种蛋白质允许癌细胞迁移到其天然配体 CXCL12 表达的其他部位,例如乳腺癌患者的骨髓中。这一证据证明了 Balixafortide 与依立替尼联合应用于转移性乳腺癌的 3 期试验以及最近用于剂量递增研究的 1 期试验是合理的。
在癌症进展和肿瘤发生中,白细胞介素(IL)的失调起着重要作用,其上调似乎与许多肿瘤有关。因此,控制其功能有利于阻止癌症发展。BNZ132-1-40 是一种 19 个氨基酸的聚乙二醇化肽,属于抗细胞因子肽家族,对白细胞介素 15(IL-15)、白细胞介素 2(IL-2)和白细胞介素 9(IL-9)有选择性地活性,这些细胞因子驱动着 T 细胞介导的疾病,包括 T 细胞大颗粒淋巴细胞白血病(T-LGLL)和人 T 淋巴细胞白血病病毒 1 型驱动的成人 T 细胞白血病(ATL)。具有螺旋构象的肽结构负责与γc 受体的直接结合,该受体存在于上述所有细胞因子中,这些细胞因子对于白血病的存活至关重要,并且缺乏强有力的活性疗法。此外,需要聚乙二醇链来增加肽的半衰期。BNZ132-1-40 对调节性 T 细胞(Tregs)的选择性,以及对 T 细胞和 NK 细胞的耐受性,通过在最近的一项 1 期临床试验中报告的健康受试者未出现对其他免疫细胞的不良影响得以证实。
ALM-201 是一种 23 个氨基酸的肽类候选药物,是 AD-01 的合成衍生物,AD-01 是一种 24 个氨基酸的肽,包含 FK506 结合蛋白类似物(FKBPL)的34 - 58序列。这种蛋白质在乳腺癌中显示出预后潜力,既可以被归类为细胞外蛋白,也可以被归类为细胞内蛋白:在第一种情况下,它作为分泌的抗血管生成蛋白,作用于在致癌情况下上调的细胞表面受体 CD44;而细胞内 FKBPL 对乳腺癌生存显示出预后活性。因此,FKBPL 参与了许多细胞过程,如细胞周期进展、信号传导和分化。与其他 FKBPL 衍生物一样,ALM201 通过 Notch 信号通路抑制乳腺癌转移,在治疗卵巢癌和实体瘤的 1 期临床试验中显示出出色的安全性。
在某些情况下,癌症生理病理学的复杂性使得无法明确地识别单一的靶蛋白来控制癌症进展,即使可以应用有效的药物治疗也是如此。例如,Fexapotide triflutate 是一种 17 个氨基酸的肽,在临床上用于前列腺癌和前列腺增生的开发。尽管其作用机制部分尚未公开,但已知 Fexapotide 通过刺激前列腺腺上皮细胞中的几个蛋白质通路(如半胱氨酸天冬氨酸蛋白酶、肿瘤坏死因子和 BCL 通路)发挥作用。它诱导选择性的细胞膜通透性、线粒体代谢停滞,并减少 RNA、DNA 的裂解和聚集,随后导致细胞碎片化和损失,引起尿道腔的减压。对动物模型的研究表明,Fexapotide 直接注射到受损组织(如膀胱、尿道、直肠、前列腺周围组织)中,不会影响相邻的未受损组织。基于所有这些证据,它已进入尿相关病理的临床试验的第三阶段。
同样,酪酪肽(YSL)是一种对原发性肝细胞癌有效的三肽化合物,目前处于临床试验的第三阶段。这种短肽具有非常高的活性且副作用很少,最初是从猪脾的水解产物中纯化的,但现在是通过化学合成获得的。为了发挥其抗肿瘤活性,YSL涉及第二信使Ca2+,它能够通过调节Ca2+/calmodulin (钙调蛋白)通路来调节细胞功能。此外,钙调蛋白与磷脂酰肌醇三激酶(PI3K)结合,增强其活性并促进细胞增殖的上调。
对于与内分泌系统功能相关的癌症类型的治疗,或者对于控制与其他化疗治疗相关的副作用,激素受体释放似乎是一个良好的靶向策略来控制和阻止肿瘤升级。艾塞那肽是一种选择性的2型胰高血糖素样肽(GLP-2)衍生物,被开发为化疗的辅助药物以减少诱导的腹泻。这种新的GLP-2和艾塞那肽合成版本显示出更长的半衰期,减少了给药频率。。该肽完成了2期临床试验。
Ozarelix是一种第四代促黄体生成激素释放激素(LHRH)拮抗剂,作用于促性腺激素释放激素(GnRH)受体,该受体控制着垂体前叶促黄体生成激素和促卵泡激素的分泌。与其他LHRH类似物一样,它是一种10个氨基酸的肽,其中包含几个D-氨基酸和大分子残基(特别是在第6位),以增强抗蛋白水解的稳定性。促性腺激素释放激素是一种十肽(EHWSYGLRPG),其N末端的三聚体片段负责激动剂活性,C末端的三聚体对于与受体的亲和力是必要的。由于主要的生物活性构象包含一个β-转角,其中两个末端靠近,因此类似物通常是通过用能够稳定所需转角的残基替换第6位的中心甘氨酸来设计的。最初,Ozarelix被设计用于刺激促性腺激素的分泌,但越来越多的证据表明,由于促性腺激素细胞脱敏和垂体受体下调,垂体激素分泌减少。事实上,作为促性腺激素释放激素(GnRH)拮抗剂,Ozarelix通过抑制垂体前叶分泌促性腺激素来降低其水平。Ozarelix完成了治疗前列腺癌的临床试验第二阶段,与其他拮抗剂相比,它具有更高的溶解性、明确的睾酮抑制效果以及不存在临床复发风险。
EP-100 是一种由 28 个氨基酸组成的合成促黄体生成激素释放激素(LHRH)天然配体衍生物,其中 18 个属于阳离子α-螺旋溶瘤肽(CLIP-71)。在多种肿瘤类型中发现了促黄体生成激素释放激素(LHRH)受体的过度表达,如前列腺、乳腺、卵巢、子宫内膜、胰腺、膀胱、结直肠、黑色素瘤和非霍奇金淋巴瘤。正因如此,它被选为治疗实体瘤和卵巢瘤的特惠靶点,EP-100 分别处于临床试验的 1 期和 2 期。LHRH 序列(HWSYGLRP)通过与细胞表面受体上的 LHRH 受体特异性结合,将溶瘤肽递送至癌细胞。EP-100 的作用模式尚不完全清楚。事实上,癌细胞带负电荷的外膜决定了这种肽对致癌组织的高选择性:在 LHRH 靶向序列结合后,EP-100 带正电荷的部分以破坏性的方式与癌细胞的外膜相互作用,导致细胞裂解和死亡。
炎症与自身免疫反应
炎症和自身免疫反应是身体防御机制的重要组成部分。它们是免疫系统识别和清除有害及外来刺激物并开始愈合过程的过程。然而,当针对自身抗原产生适应性免疫反应或存在促炎分子的上调招募时,免疫效应机制通常无法完全清除抗原/促炎分子,因此会发生持续反应。从这个角度来看,开发了能够干扰失调的身体防御机制的基于肽的治疗方法。
Forigerimod是一种合成的 21 个氨基酸的线性肽,在对应丝氨酸 11 位点磷酸化,由 ImmuPharma 公司生产,并根据小核核糖核蛋白 U1 - 70设计。正如其商品名所暗示的,Lupuzor 用于治疗系统性红斑狼疮(SLE),这是一种慢性疾病。一种危及生命的自身免疫性炎症性疾病,影响多个器官,如皮肤、关节、肾脏、血细胞、大脑、心脏和肺部。由于这种病理的高相关性,Lupuzor™ 获得了美国食品药品监督管理局(FDA)的快速通道认定,这加快了该药物的批准过程。目前,Lupuzor™ 的作用机制尚未完全阐明;然而,多项研究表明,它具有致耐受和免疫调节作用,能在内源性肽存在的情况下抑制 T 细胞的反应性。
另一方面,对于系统性炎症反应综合征(SIRS)的治疗,由 Exponential Biotherapies 公司生产的合成四肽 EA-230 的静脉输注目前正在研究中(2 期)。该序列是基于内源性肽人类绒毛膜促性腺激素(hCG)β亚基中包含的β环设计的。SIRS 病理是由一系列促炎因子(如 IL-6)的上游作用引起的,这种失调的炎症反应往往会导致组织损伤、一个或多个器官系统衰竭以及高死亡率。尽管 EA-230 的作用机制仍不清楚,但生物学证据已经表明,激素环境的变化如何在抗炎反应中发挥核心作用。
另一种用于治疗炎症性疾病的肽是醋酸Difelikefalin,这是一种由卡拉治疗公司生产的合成5-mer linear D-peptideproduced,其设计起始于内源性肽双诺啡A的序列。因此,醋酸Difelikefalin是κ-阿片类受体(KOR)的激动剂,用于治疗慢性肾病相关性瘙痒(CKD-aP)或尿毒症性瘙痒,并且2020 年的临床试验已完成 3 期试验。。慢性肾脏病相关性瘙痒症(CKD-aP)是一种极其痛苦的病症,在接受透析的患者中超过 60%会出现这种情况,尽管其发病机制仍不完全清楚,但阿片类药物失衡已被确定为这种病症出现的一个可能原因。由于 KOR(在外周和中枢神经系统中)的普遍性,醋酸Difelikefalin被设计为一种选择性激动剂,避免穿透中枢神经系统,旨在通过激活抗瘙痒的 KOR 系统来最小化瘙痒,同时不会引起中枢神经系统的副作用。
谈到影响呼吸系统的炎症,可以提到LSALT肽,这是一种由Arch Biopartners 开发的合成16个氨基酸的线性肽,目前仍处于 2 期临床试验阶段,用于治疗急性呼吸窘迫综合征(ARDS)。这种炎症的一个显著特征是中性粒细胞从血液被招募到炎症组织中。在这种情况下,二肽基肽酶 - 1(DPEP1)是一种锚定膜蛋白,已被确定为肺和肝内皮细胞中中性粒细胞捕获的主要黏附受体,独立于其酶活性。因此,中性粒细胞向多个器官的不当招募会导致多器官功能障碍(如肺功能障碍)。为了防止这些健康功能障碍,几个实验模型已经表明,LSALT 肽与 DPEP-1结合,不是抑制其酶活性,而是阻止中性粒细胞的捕获和炎症反应。
在急性呼吸窘迫综合征(ARDS)的治疗范围内,Avipdtadil 是一种由 NeuroRX 和 Relief Therapeutics Holding 开发的合成 28 个氨基酸的线性肽,其设计灵感源自内源性血管活性肠肽(VIP)的结构。这种内源性蛋白质与肺动脉高压(PH)的形成有关,PH 是一种由小动脉血管收缩和结构重塑引起的进行性血管疾病,会导致呼吸困难、疲劳、咳嗽、胸痛、心悸、外周水肿、晕厥、右心衰竭和死亡。此外,如果不进行治疗,PH 在 ARDS 患者中的发病率会在 5 年内减半,生存率降低。作为 VIP 类似物,Avipdtadil 通过与 G 蛋白偶联受体(VPAC1、VPAC2 和 PAC1)结合,诱导肺血管扩张,并表现出抗炎特性。事实上,Avipdtadil 气雾剂仍在 2/3 期临床试验中,用于治疗 ARDS,导致小而暂时的但显著的选择性肺血管扩张,以及每搏输出量和混合静脉血氧饱和度的改善。
Larazotide是9M生物制药公司(9M Biopharma Inc., 2021)正在研究的药物,是唯一一种用于乳糜泻辅助治疗的治疗候选药物,已推进到 3 期临床试验阶段,它能与无麸质饮食协同作用,最大程度地减轻症状。这种八肽源自霍乱弧菌分泌的闭锁小带毒素,是已知唯一的生理性细胞间紧密连接的调节剂,也是小肠黏膜免疫反应调节中的关键因素。
谈到身体更具体的部位,耳部和眼部炎症的治疗尤其引人关注。
Brimapitide 是一种由 Auris Medical 开发的合成右旋甘氨酸31个残基的线性肽,其设计基于胰岛脑1(IB-1)的20个残基序列和HIV TAT 蛋白的 10 个残基转录激活序列(TAT)的组合,这使得它能够穿透细胞。它与 IB-1 的相似性使这种肽成为 c-JunN 氨基末端激酶(JNK)的选择性抑制剂,JNK 是一种普遍存在的细胞内酶。其抑制作用可防止转录复合物的形成,并阻止细胞凋亡途径的进一步进展或基因的激活,这些基因编码炎症分子(如细胞因子)。因此,Brimapitide 正在研究中(3 期临床试验),分别作为生物相容性鼓室内透明质酸凝胶和眼科溶液用于治疗急性感音神经性听力损失(ASNHL)和眼部炎症。
关于眼部炎症的治疗,值得一提的是由RegeneRX Biopharmaceuticals公司生产的Timbetasin,这是一种合成的43个氨基酸的线性肽,其作用类似于内源性肽Thymosin β4,但N末端乙酰化有所不同。因此,Timbetasin被用于治疗中重度干眼症,因为它具有抗炎作用。干眼症是一种慢性眼表疾病,导致视力障碍,影响生活质量,并且与结膜和泪液中炎症细胞因子水平的增加有关。一般来说,内源性Thymosin β4在各种动物模型中促进皮肤、眼睛、心脏和神经系统的伤口修复和再生;此外,它还调节多种信号分子的表达,参与下调炎症趋化因子、细胞因子和金属蛋白酶的转录因子。出于这个原因,Timbetasin 作为滴眼液局部补充剂目前正在研究中(3 期临床试验),以促进眼表愈合、增加角膜上皮细胞迁移,并降低角膜促炎细胞因子水平。
最后,对于骨组织炎症的治疗,OrthoTrophix 正在研究一种名为 TPX-100 的合成 23 肽,其结构目前尚未公开。这种肽由 OrthoTrophix 生产,并从基质细胞外磷酸糖蛋白(MEPE)开始设计,MEPE 是一种属于小整合素结合配体 N 连接糖蛋白(SIBLING)家族的蛋白质。有证据表明,这些蛋白质在骨和牙本质的矿化过程中发挥关键作用,但其作用机制尚不清楚。研究表明,随着时间的推移,膝关节的形状会发生单向且不可逆的变化,骨关节炎膝关节的变化率远高于正常膝关节。这种加速的骨形状突变比软骨退变开始得早得多,并且迄今为止似乎是骨关节炎(OA)发病的最可靠结构标志物。即使其作用机制仍未公开,TPX-100 已完成治疗膝关节骨关节炎的 2 期临床试验,诱导正常胫股关节透明软骨的再生,并强调了胫股关节软骨的稳定与关键膝关节功能之间的统计学显著相关性。
遗传和激素性疾病
基因疾病可能是由单个基因的突变(单基因疾病)、多个基因的突变(多因素遗传疾病)、基因突变与环境因素的结合,或者染色体损伤引起的(国家人类基因组研究所 NHGRI,2021 年)。另一方面,激素疾病可能是由于腺体本身的问题,或者由于下丘脑 - 垂体轴(下丘脑和垂体之间激素信号的相互作用)提供过多或过少的刺激而导致的。在这种情况下,基于肽的治疗既可以作为缺乏内源性分子的补充(基因疾病),也可以作为干扰导致其生产失调的机制的介质(激素疾病)。
HTL0030310 是由SoseiHeptares 公司生产的一种新型合成肽,该公司最近完成了针对健康受试者的安全性及耐受性的临床试验的第一阶段。尽管 HTL0030310 的结构仍未公开,但SoseiHeptares 公司将其设计为选择性 SSTR5(生长抑素 5)受体激动剂,用于治疗内分泌系统疾病,调节垂体腺瘤(良性肿瘤)过度释放激素的情况。垂体激素血浆水平大幅升高会导致许多严重疾病,包括库欣病,这是一种由皮质醇激素过度产生引起的使人虚弱的内分泌系统疾病。
另一方面,沃索利肽是一种由BioMarin制药公司生产的合成39个氨基酸的环肽,目前正处于治疗人类最常见的短缩畸形——软骨发育不全的临床试验第三阶段。这种疾病是由成纤维细胞生长因子受体3(FGFR3)的常染色体显性突变引起的,该基因持续激活丝裂原活化蛋白激酶(MAPK),从而抑制软骨内骨化。由于C型利钠肽(CNP)及其受体利钠肽受体2(NPR2)是软骨内骨化的强大刺激物,因此观察到持续静脉输注外源性C型利钠肽可恢复受损的骨生长。沃索利肽被设计为C型利钠肽的环状类似物,以增加其与内源性形式的半衰期相比的半衰期。
糖尿病、肥胖症、短肠综合征与高胰岛素血症,胰高血糖素的遗患
在过去几十年里,一系列源自肠道内分泌系统的合成肽被确定为治疗代谢性疾病(如2型糖尿病和肥胖症)的有希望的方法。这些疾病仍然是全球健康问题,不断增加,已知会降低生活质量并导致严重并发症。2型糖尿病的治疗涉及针对体重的药物,同时维持血糖控制。
主要的合成治疗性肽是通过模仿源自前激素前胰高血糖素代谢裂解的序列设计的。这种158个氨基酸的小蛋白质在胰腺、肠道和中枢神经系统中被前激素转换酶进行特定的翻译后加工,从而产生较短的片段(图 5)。其中,胰蛋白酶抑制肽(GRPP)、胰高血糖素、中间肽-1(IP-1)和中间肽-2(IP-2)、主要胰高血糖素前体片段(MPGF)、催吐激素(OXM)、胰高血糖素样肽-1(GLP-1)和胰高血糖素样肽-2(GLP-2)在血糖控制、能量平衡和肠道稳态中发挥着重要作用,因此被提议作为药物设计的模型。
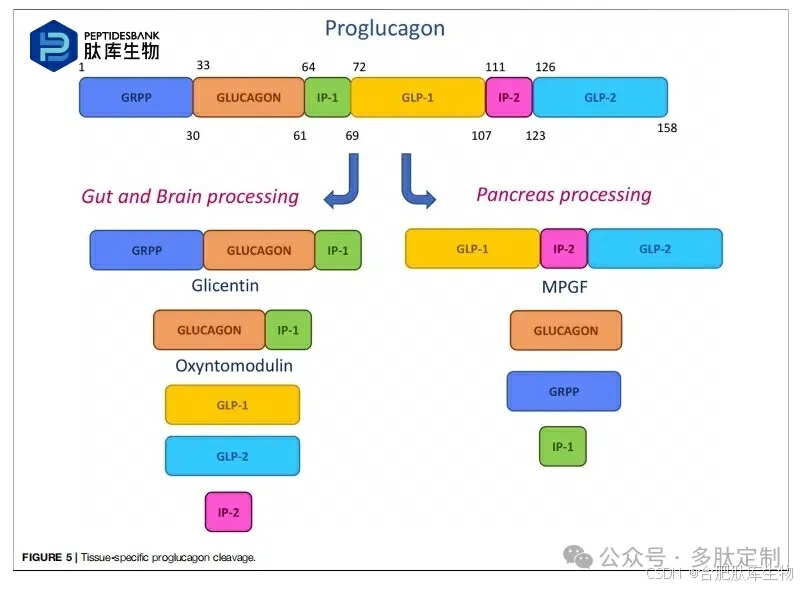
胰高血糖素样肽-1受体靶向肽
药物作用的主要生物靶点之一是胰高血糖素样肽 - 1 受体(GLP - 1R),它属于 B 类 G 蛋白偶联受体(GPCR),深度参与胰岛素的分泌和稳态。与其他 G 蛋白一样,GLP - 1R 的特征是具有七个跨膜α-螺旋,其间隔有三个细胞内环和三个细胞外环。
GLP-1R 的激活与胰高血糖素样肽 1(GLP-1)的分泌有关,GLP-1 是一种肠促胰岛肽,在进食后于肠道中释放。GLP-1 存在两种形式:GLP-1(7-37) 和 GLP-1(7-36) 酰胺,后者在进食后于循环中更丰富。这些肽与 GLP-1R 相互作用,通过受体信号传导、通过环腺苷酸(cAMP)的产生来增强胰岛素分泌并使血糖水平正常化。当调节作用完成时,GLP-1 会通过蛋白水解失活。除了这种主要作用外,GLP-1 及其激动剂还可以减少胰高血糖素的分泌、减缓胃排空并降低能量消耗,从而延长对肥胖症的治疗效果。GLP-1R/GLP-1 系统的关键作用体现在近期为开发有效的 GLP-1R 肽类激动剂以用于 2 型糖尿病(T2DM)的药理治疗所做的努力中。
在发现Gila monster蜥蜴的毒液中含有艾塞那肽-4 后,该领域取得了重大进展。艾塞那肽-4(也被称为艾塞那肽,即其合成形式)是一种 39 个氨基酸的肽,能抵抗二肽基肽酶 4(DPP-4),由于其序列中有一个甘氨酸残基,其药代动力学性能优于 GLP-1。艾塞那肽于 2005 年获得美国食品药品监督管理局(FDA)批准,每日注射两次。目前,治疗 2 型糖尿病(T2DM)和肥胖症的最佳药物是利拉鲁肽和最近口服的司美格鲁肽。这两种分子促进了对具有更优药理特征的 GLP-1 类似物更好的研究。
为了满足这些治疗需求,对艾塞那肽序列的进一步修改导致了缓释化合物:例如,由 PegBio 开发的聚乙二醇化艾塞那肽 PB-119 就是一个例子,它通过对艾塞那肽中单个氨基酸的修饰而获得,用Lys1-ξ-NH 2 取代Ser39with 半胱氨酸。即使其作用机制与艾塞那肽相同,聚乙二醇化类似物 PB-119 降低了在肾脏中的排泄率,其增加的分子量对 DPP-4 造成了空间位阻,延长了半衰期,并允许每周进行一次皮下注射剂量方案。江苏汉索制药也通过开发 GLP-1R 激动剂诺和格列汀(一种 40 个氨基酸的艾塞那肽类似物)采用了类似的理念,以改善药代动力学和抗 DPP-4 的能力,目前该药物正处于 2 期试验阶段,作为皮下注射剂。从结构上讲,在 2 位存在非天然 Aib(α-氨基异丁酸)残基应能保证更好地抵抗酶水解,而与棕榈酸在 2 位的缩合增强了亲水性。
GLP-1R 激动剂的名单中还包括由帝国理工学院开发的 G3215(结构未公开),该药物于 2019 年完成了其 1 期试验。
如上所述,GLP-1R 激动剂肽类越来越受到关注,有几种药物已进入临床阶段。相比之下,对 GLP-1R 拮抗剂的研究投入较少。然而,Eiger BioPharmaceuticals 正在开发 Avexitide 用于治疗减肥术后低血糖(PBH),这是一种高胰岛素性低血糖(HH)的形式,旨在作为一种新的液体形式的 Exendin 9−39,用于皮下注射。PBH 是一种罕见的疾病,目前没有获批的药物治疗,其由胰岛素分泌异常增长引起,这归因于餐后分泌的过度增加以及餐诱导的 GLP-1 释放量的大幅增加。因此,GLP-1R 拮抗作用可能是管理这种病症的潜在治疗策略。Avexitide 是一种 31 个氨基酸的肽,可特异性地靶向并阻断 GLP-1R,使胰岛素分泌正常化,从而减少餐后低血糖。从一项研究中发布的数据显示最近的 2 期临床试验表明,艾塞那肽在低血糖发生率方面产生了持续的改善,并保持良好的耐受性。
胰高血糖素样肽-1受体/GPCR 胰高血糖素受体靶向肽
GLP-1R 激动肽在临床上的成功促使人们寻找能够将 GLP-1 的益处与其他关键肠道激素的益处相结合的药物,以便用单一的分子实体靶向多个信号通路。此外,这种新兴的双重药理方法可能比单一疗法能产生更强的代谢作用,许多候选药物正在临床试验中进行评估。
葡萄糖稳态由胰岛素和胰高血糖素的协同作用调节。虽然胰岛素及其治疗应用得到了广泛研究,但胰高血糖素激素(由胰腺α细胞分泌)在过去几十年中才受到关注。其主要作用是对胰岛素产生反调节作用,通过靶向 B 类 GPCR 胰高血糖素受体(GCGR)诱导肝糖原分解和糖异生。在治疗低血糖的治疗方法中使用天然胰高血糖素并非易事,因为其在生理制剂中稳定性极低,而且在复配后几个小时内就极易聚集。因此,在治疗低血糖方面,开发天然胰高血糖素肽的新类似物/激动剂是必要的。Zealand Pharma 开发了 Dasiglucagon(NDA),它将于 2021 年第一季度以 Zegalogue® 的形式推向市场,这是一种通过单针注射笔应用的即用型制剂。为避免聚集问题和原纤维的形成,并使其在水溶液中具有物理和化学稳定性,对天然胰高血糖素的设计引入了七处取代,在 29 个氨基酸序列中插入了一个 Aib 残基。
另一方面,胰高血糖素能增加能量消耗,并起到食物摄入调节剂的作用,这使得其类似物在调节 2 型糖尿病(T2DM)和肥胖症中的血糖方面也颇具吸引力。GLP-1 和胰高血糖素具有高度的序列同源性:这一方面促使开发出多种具有综合药理特征的分子,以双重激动机制同时作用于 GLP-1R 和 GCGR,在这种机制中,血糖和体重的控制伴随着脂肪组织的分解和食欲抑制。天然肠道激素肽 Oxyntomodulin(OXM)在进食后被分泌,具有抗糖尿病作用,并通过激活 GLP-1R 和 GCGR 来驱动体重减轻,作用于食欲和饱腹感,尽管其活性低于单独的 GLP-1 和胰高血糖素。因此,Oxyntomodulin 已被选为开发 GLP-1R/GCGR 双重激动剂的模型。
在这个类别中,OPKO Health 公司的 Pegapamodutide(OPK880033)完成了每周一次给药治疗 2 型糖尿病(T2DM)的 2 期临床试验。这种候选药物是 OXM 的 PEG 化类似物,由于在Cys38and Cys39处存在两个 PEG 基团,可防止酶降解。序列中存在的两个非天然 Aib 残基也提供了对二肽基肽酶 - 4(DPP - 4)的抗性。特别是,Aib2增强了对 GLP1 - R 的效力和选择性。
此外,阿斯利康的子公司 MedImmune 正在开发 Cotadutide(MEDI0382),目前处于 2 期试验阶段,作为每日一次的皮下治疗,用于治疗患有 2 型糖尿病(T2DM)的超重或肥胖患者。这种新型双 GLP-1R/GCGR 激动剂旨在通过联合作用于食欲和能量消耗来改善血糖控制并促进体重减轻。从结构上讲,MEDI0382 是一种 30 个残基的合成线性肽,设计为 OXM 类似物,其中催产素的序列在 C 末端被去除了七个残基,同时剩余的七个氨基酸中有七个被改变。通过增强对肽酶降解的稳定性实现了其延长半衰期:将 Gln20和 Gln24替换为不易脱氨的残基,同时将 Arg17替换为 Glu 以减少蛋白水解。此外,通过在 Lys10处通过γ-谷氨酸基团插入棕榈酸侧链促进了其与白蛋白的可逆结合。通过这种方式,这种抗降解肽能够发挥最优平衡的双重激动作用,在临床试验中观察到患者的体重减轻效果优于单独使用 GLP-1R 的效果。
另一种长效的 GLP-1R/GCGR 双激动剂 BI456906 正由泽兰制药与勃林格殷格翰合作开发。这种肽是天然 OXM 的类似物,目前正在进行 2 期研究,用于治疗 2 型糖尿病(T2DM)和肥胖症。由于针对这种病理使用 GLP-1R/GCGR 双激动剂的最新成功方法,BI456906 作为非酒精性脂肪性肝炎(NASH)(非酒精性脂肪性肝病(NAFLD)的最严重形式)的治疗药物的疗效也有望实现。
在这个领域,ALT-801 作为由 AltImmune 设计的一种新型基于肽的双重 GLP-1R/GCGR 激动剂,用于治疗非酒精性脂肪性肝炎(NASH)引起的肥胖和代谢功能障碍。正如在该疾病的临床前模型中所观察到的,与司美格鲁肽相比,ALT-801 引起了显著的体重减轻,相应地,该公司最近宣布开始进行 1 期临床试验。
其结构基于一个 29 个氨基酸的序列,部分模仿 GLP-1 以改善减重效果,并模仿胰高血糖素以恢复代谢功能。插入 Aib 残基旨在防止蛋白水解降解,同时在 Lys17 侧链上引入由 D-葡萄糖苷与亚甲基 C18 链连接的 EuPort 脂多糖表面活性剂,以提高胃肠道耐受性。此外,Glu16 和 Lys20 侧链被连接以形成内酰胺环,设计为螺旋稳定剂以增强效力。这些结构特征使得它与血清白蛋白有高亲和力,适合患者每周一次皮下注射。最后,LY3305677(OXM3,IBI362)是一种未公开的肽类双 GLP-1R/GCGR 激动剂,礼来公司报告称其为第三代催产素的类似物,目前正在 2 期临床试验中进行评估。
胰高血糖素样肽 - 1 受体/GIPR靶向肽
治疗糖尿病和肥胖症的另一种策略是利用对胰高血糖素样肽受体 GLP-1R 和 GIPR 的激动作用。GIPR 是天然肠道激素葡萄糖依赖性促胰岛素多肽(GIP)的天然靶点,由 K 细胞在进食后分泌。这种促胰岛素肽可以像 GLP-1 一样刺激胰岛素的分泌,但对胰岛素分泌的调节效果较弱。关于受体-配体相互作用的研究指出,天然肽的 N 末端在 GIPR 激活中起关键作用。这一部分与跨膜受体的结合位点相互作用,而中心残基与受体的细胞外结构域结合。因此,对 N 末端部分的调整可以调节肽的激动作用或拮抗作用。
因此,GIP 激动作用已被确定为治疗 2 型糖尿病(T2DM)的一种药物策略,尽管它似乎不如 GLP-1 激动作用的相关性大。在过去几年中,被称为“双促胰岛素受体激动剂”的合成双促胰岛素受体激动剂得到了研究,实际上有一些候选药物正在临床开发中。最先进的 GLP-1R/GIPR 激动剂是礼来公司(Eli Lilly)的 Tirzepatide(LY3298176),目前正处于 3 期试验阶段。Tirzepatide 被制成一种合成的 39 个残基的线性肽,与天然 GIP 有 19 个残基相同。该肽序列在 2 位和 13 位包含了酰胺化的 C 末端和两个非天然 Aib 残基。此外,该结构通过一个亲水连接子与 Lys20残基相连,与一个 20 碳脂肪二酸部分相连。这种策略旨在促进白蛋白结合,将 Tirzepatide 的半衰期延长至约 5 天,从而能够实现每周一次的皮下给药方案。在 1 期和 2 期临床试验中,与 GLP-1 激动剂度拉古肽相比,Tirzepatide 在血糖控制和减重方面的临床疗效、安全性和耐受性有了显著改善。GIPR 的结合亲和力与天然 GIP 相当,然而观察到 GLP-1R 的亲和力比天然 GLP-1 低五倍。
值得一提的是,由史克公司与武田制药合作开发的双激动肽 SCO-094也属于这类候选药物。该分子正在进行长效和口服制剂的 1 期测试,具有广泛的应用潜力,包括糖尿病、肥胖症和非酒精性脂肪性肝炎。
最后,由 Carmot Therapeutics 开发的双激动剂 CT-868 将于 2021 年上半年进入 2 期试验,有望成为治疗糖尿病、肥胖症和非酒精性脂肪性肝炎(NASH)的良好药物。CT-868 双激动剂候选药物是通过 Chemotype Evolution 技术发现的,它是一种肽类小分子混合化合物,能够模拟天然 GLP-1 激素。
基于双靶向肽的有前景的结果,一些同时靶向 GLP-1、GCGR 和 GIPR 的三元激动剂已经被开发出来。来自礼来公司的肽 LY3437943 是这一新型肽类的第一个成员,旨在对 2 型糖尿病(T2DM)和肥胖症的治疗有效。这种肽的结构仍未由原研公司公开。
Y受体2型靶向肽
其他针对肥胖及相关病理的潜在疗法涉及胰高血糖素样肽 - 1(PYY)和淀粉样肽激动剂。
肽YY(PYY)是一种肠道激素,由肠内分泌L细胞与GLP-1和催吐激素共同分泌,以响应进食。尽管它存在两种主要形式,即全长PYY1-36和PYY3-36,但后者是最常见的具有生物活性的循环形式。PYY3-36是一种34个氨基酸的肽,在餐后由肠道释放。它的分泌通过作用于下丘脑弓状核中的神经肽Y受体2型(Y2R)来激活食欲抑制和减少食物摄入,Y2R是神经肽Y受体家族的一员,属于GPCR家族。
早期的临床研究表明,在肥胖患者中注射 PYY3-36 可抑制食物摄入,耐受性良好,副作用有限。然而,由于天然 PYY 半衰期短(8 分钟)会影响临床稳定性,人们计划了各种方法来增加其抗蛋白水解失活的能力。在这种情况下,替代 PYY 的给药方式是一个挑战。最近,Gila Therapeutics 开发了 GT-001,这是一种 PYY3-36 类似物,已开始口服舌下制剂的 1 期试验。诺和诺克的研发线中还包括一种调节食欲的新型 PYY 类似物,即 PYY-1875,其结构仍未公开。这种肽目前正在进行 1 期试验,旨在每周注射一次以供皮下注射治疗。
淀粉样肽是一种 37 个氨基酸的肽类激素,主要在胰岛β细胞中产生,并在进食期间与胰岛素共同分泌。淀粉样肽作为一种饱腹感信号的作用机制已得到充分确立;它通过与大脑特定区域的人类淀粉样肽受体(AMY)结合,减少食物摄入量和餐后胰高血糖素分泌。AMY 亚型是GPCR,由降钙素受体(CTR)和三种受体活性调节蛋白(RAMP)之一组成。由于其作用机制,淀粉样肽类似物是作为抗肥胖药物的新型研究靶点,并且已经探索了几种方法(PEG 化、糖基化或白蛋白结合)来延长其半衰期并减少给药频率。诺和诺德目前正在进行 AM833 的 2 期试验,这是一种长效的酰化人类淀粉样肽激素的类似物,每周皮下给药一次。AM833 已在 1 期临床试验中与 GLP-1 类似物司美格鲁肽联合使用进行了评估。
胰高血糖素样肽-2受体靶向肽
胰高血糖素样肽-2受体(GLP-2R)是GPCR超家族成员,在胃肠道中表达,属于七跨膜受体家族,与糖皮质激素受体(GCGR)和胰高血糖素样肽 - 1 受体(GLP - 1R)密切相关。通过与胰高血糖素样肽 - 2(GLP - 2)结合,GLP - 2R 直接通过响应配体激活刺激细胞增殖来抑制细胞凋亡并促进肠道生长。GLP - 2 是一种 33 个氨基酸的激素,由肠内分泌的肠道 L 细胞响应营养物质摄入而释放。
尽管 GLP-2 介导其作用的机制仍不完全清楚,但它对于刺激肠道生长、增加吸收、促进愈合以及维持上皮完整性而言,在正常人以及因大规模肠切除(短肠综合征,SBS)而肠衰竭的患者中,都是一个重要的调节因子。事实上,SBS 患者可能存在餐后 GLP-2 分泌受损的情况,而这是实现最佳肠道适应所需要的。由于 GLP-2 会被 DPP-4 裂解,导致其循环半衰期非常短(约 7 分钟),因此将其作为治疗药物的使用受到限制。针对 SBS,目前正在研发具有更长半衰期和减少清除率的具有药理活性的 GLP-2 类似物,包括 3 期药物阿普拉格鲁肽(VectivBio)和格列格鲁肽(Zealand Pharma)。
格列帕肽是一种高效的 GLP-2R 选择性 39 肽,具有长效作用,血浆有效半衰期约为 50 小时,可每日少于一次给药。此外,它通过现成自动注射器设备提供了更方便的给药形式,无需从冻干粉末中复配,并且在注射部位形成皮下储库,可缓慢释放到循环中。格列帕肽与天然 GLP-2 的不同之处在于插入了一个由六个赖氨酸残基组成的 C 末端尾链,改变了电荷状态,有助于溶解性和物理化学特性。通过改变五个氨基酸优化了 N 末端区域,以改善药代动力学和效力特性,并在内部区域用丙氨酸替换其他四个残基,最终导致全局九个氨基酸取代。
阿格拉鲁肽是另一种高选择性、强效的 GLP-2R 激动肽,由 33 个氨基酸序列组成,其分子结构旨在保持最佳药理活性,同时与天然 GLP-2 或其他 GLP-2 类似物相比延长半衰期。具体而言,天然 GLP-2 在 2、10、11 和 16 位分别被甘氨酸、亮氨酸、D-苯丙氨酸和亮氨酸修饰,并在 C 末端进行酰胺化。这些少量的氨基酸取代转化为优异的药代动力学特征,导致清除率极低,血浆蛋白结合率高,从而能够在皮下给药后实现长久的体内消除半衰期(30 小时),无需缀合或其他肽修饰。此外,阿格拉鲁肽对 DPP-4 表现出良好的稳定性,使得每周只需治疗一到两次成为可能。
在干燥综合征(SBS)患者中,对天然胰高血糖素样肽(GLP)肽的作用进行了研究,其作用方式包括作为选择性激动剂(GLP-1R 或 GLP-2R)或作为双 GLP 激动剂联合疗法。与单独使用每种药物相比,后者显示出更优的疗效和更好的患者预后。这一原理支持使用双 GLP 受体共激动作用来治疗代谢和胃肠道疾病,即使临床研究仍在等待确认其联合治疗的效果。遵循这一方法,泽兰制药目前正在开发地那谷肽(Dapiglutide),此前被称为 ZP7570。尽管其结构仍未公开,但该公司宣布,这种分子在 2020 年完成了 1 期试验,在健康志愿者中显示出良好的安全性和耐受性,血浆半衰期允许每周给药一次。
心血管系统与高血压
心血管疾病(CVD)是全球主要的死亡原因之一,每年估计夺走179万人的生命。根据世界卫生组织(WHO)的数据,五分之四的心血管疾病死亡是由于心脏病发作和中风,其中三分之一发生在7岁以下的儿童身上(WHO,2021)。近年来,具有独特生物活性和代谢的蛋白质和肽类成功地吸引了研究人员的关注,作为心血管和高血压疾病的替代治疗方法,其中一些仍在临床研究中等待批准。
Ularitide是一种心房肽激动剂,由专门治疗心血管疾病的私营生物制药公司Cardiorentis开发,用于急性心力衰竭和肾衰竭的静脉治疗,并于2018年完成了3期临床试验。乌拉立肽是人类内源性利钠肽尿激肽的化学合成形式,由肾脏产生,旨在调节液体平衡和钠稳态。这种含有32个氨基酸的肽与含28个氨基酸的心房利钠肽(ANP)具有相同的序列,只是在N末端添加了四个氨基酸。与受体结合的ANP的晶体结构显示,该复合物包含两个NPR-A分子与一个ANP分子结合,由于ANP分子没有内部对称性,与受体的结合是不对称的,通过每个NPR-A单体中存在的两个不同结合位点发生。乌拉立肽主要与钠肽受体 A(NPR-A)的细胞外结构域结合,NPR-A 在心脏、肾脏和其他器官中表达,并激活该受体的细胞内鸟苷酸环化酶结构域。鸟苷酸环化酶催化鸟苷酸 5‘ - 三磷酸转化为环鸟苷酸 - 3’,5' - 单磷酸(cGMP),通过平滑肌细胞的血管舒张引起血管扩张,并通过抑制钠重吸收引起尿钠和利尿作用。在治疗中风相关疾病方面,富含精氨酸的阳离子肽(CARPs)代表了一类新兴的具有多模式细胞保护作用的神经保护剂。其中,NA-1(最初名为 TAT-NR2B9)是 NoNO 公司开发用于治疗中风、创伤性脑损伤和蛛网膜下腔出血的先导化合物,目前正处于 3 期临床试验阶段。NA-1 是一种 20 个氨基酸的肽,其序列为(KLSSIESDV),源自细胞内末端羧基N-甲基-D-天冬氨酸(NMDA)受体NR2B亚基蛋白的区域,与富含阳离子精氨酸的细胞穿透肽TAT(YGRKKRRQRRR)融合,有助于穿过血脑屏障。NR2B9 9-肽被选择来抑制与NR2B亚基结合的突触后密度蛋白-95(PSD95)衔接蛋白,从而阻断与NMDA受体过度刺激相关的下游细胞信号传导。
CN-105 是一种小型的 5 个氨基酸的载脂蛋白 E(apoE)模拟肽,富含精氨酸残基,源自载脂蛋白 E 的受体结合区域,目前由 AegisCN(前身为 CereNova)开发。这种肽正处于治疗出血性中风和脑出血的 2 期开发阶段。CN-105 是基于亲神经保护性肽 COG133 中存在的氨基酸开发的,COG133 包含载脂蛋白 E 内的肝素结合和低密度脂蛋白受体结合域。载脂蛋白 E 是一种多功能的多肽,可减少神经炎症,并在缺血性和创伤性脑损伤后介导适应性反应。然而,完整的载脂蛋白 E 全蛋白无法穿过血脑屏障(BBB),限制了其治疗潜力。CN-105 具有增加中枢神经系统渗透性的优势,并且在实验性脑出血中显示出疗效,导致实验性缺血性中风后的功能和组织学结果得到改善,并减少小胶质细胞活化。
目前仍在临床试验中用于治疗心血管疾病的通过蛋白质-蛋白质相互作用的最后一种肽是Pemziviptadil(PB1046)。PB1046是一种新型、皮下注射的血管活性肠肽(VIP)类似物,由PhaseBio Pharmaceutical开发,目前正在进行2期试验,用于治疗肺动脉高压(PAH)。VIP是一种28个氨基酸的肽激素,能够激活肺血管中的VPAC1和VPAC2受体,已被证明能够松弛肺血管平滑肌,中和肺血管收缩剂,并抑制细胞增殖。VIP的半衰期较短,使得这种肽作为药物制剂不太实用,因此需要对其进行修改,以使其具有治疗效果。PB1046是一种与弹性蛋白类似多肽(ELP)生物聚合物连接的29个氨基酸的肽。ELP部分包含与弹性蛋白相关的结构肽片段。这种修饰的序列有助于改善重要的特性,如吸收率和循环半衰期。
神经退行性疾病
神经退行性疾病是基于退行性细胞变化的一个持续过程的结果,随着时间的推移,病情会日益恶化。致病蛋白质的聚集、线粒体功能障碍、氧化应激、转录功能障碍和细胞凋亡在帕金森病(PD)、阿尔茨海默病(AD)和肌萎缩侧索硬化症(ALS)等神经退行性疾病的发生发展中起着重要作用。迄今为止,还没有新的疾病修正疗法被证明能为患有这些毁灭性疾病患者带来显著益处。因此,市场上越来越需要早期诊断以及发现能够选择性地与特定靶点结合的新药。
Davunetide(NAP,AL-108)是一种通过鼻腔给药的8-肽片段,源自依赖活性神经保护蛋白(ADNP),目前正处于治疗进行性核上性麻痹(PSP)的1期临床试验阶段。Davunetide通过促进微管稳定具有神经保护、神经营养和认知保护特性,具有临床前证据。构成微管骨架的蛋白质微管蛋白已被证明是NAP的目标,最近报道称NAP能增强微管与微管相关蛋白之间的相互作用,并在锌中毒条件下促进微管聚合。NAP还通过阻断小胶质细胞激活来降低神经毒性因子和促炎因子(如一氧化氮(NO)和肿瘤坏死因子(TNF))的水平。微管末端结合(EB)蛋白已被确定为NAP的目标,通过其SIP结构域的相互作用。达文内肽与其受体的结合最近通过将EB3与EB1-EB3复合物与另一种肽配体(MACF)的结构比对进行预测。
NLY01(Neuraly 公司)是一种聚乙二醇化的长效艾塞那肽-4类似物。在 1 期研究中,NLY01 耐受性良好,其半衰期比作用时间较短的 GLP-1R 激动剂高三倍,而后者往往受副作用的限制。2020 年,宣布批准一项新药临床试验申请(IND),以启动 NLY01 在阿尔茨海默病患者中的 2b 期临床试验以及在帕金森病患者中的 2 期试验。
在之前的一项研究中,结果表明 NLY01 通过良好的血脑屏障渗透,与上调的小胶质细胞源性谷氨酸受体 1(GLP-1R)结合,在包括帕金森病在内的神经退行性疾病动物模型中阻断小胶质细胞的病理激活。
重症肌无力(MG)是另一种严重的退行性疾病,是一种慢性自身免疫性疾病,其中自身抗体攻击神经肌肉接头中的特定蛋白质,导致肌肉无力和疲劳。Zilucoplan是由UCB制药公司开发的一种小型的合成15-环大环肽,目前正处于MG的3期临床试验阶段,它作为终末补体蛋白C5的强效抑制剂,具有潜在的抗炎和细胞保护作用。通过皮下给药,Zilucoplan与补体蛋白C5结合,阻止C5裂解为C5a和C5b,并防止C5b依赖的膜攻击复合物(MAC)的组装。Zilucoplan还抑制C5b与C6之间的相互作用,从而进一步阻止MAC的组装。
最后但同样重要的是,GM604(GM6,Alirinetide)是一种由 Genervon 生物制药公司开发的阳离子线性肽类药物,目前正处于治疗ALS疾病的2期临床试验中。该肽由6个氨基酸组成,代表内源性33-单肽“运动神经营养因子”(MNTF1)的一个单位。临床前试验表明,MNTF调节中枢神经系统的生物学功能,包括神经元分化、轴突再生、神经再生、炎症和凋亡,提供神经保护和神经再生治疗效果。预计GM604具有复杂的作用机制,可能涉及刺激多种受体、信号级联和下游基因表达反应。GM604不选择性地与单一靶标相互作用,而是与多种途径相关的多个受体相互作用。这些包括胰岛素受体,Notch受体1到4和平滑卷曲类受体。GM6对这些和其他受体的刺激和激活与数千个基因的表达有关,这些基因导致微管稳定性、突触传递和轴突引导。
细菌和病毒感染
由包括细菌、病毒、真菌和寄生虫在内的许多病原体引起的传染病在全球范围内极为常见,尽管市场上已有大量抗生素和抗病毒药物。必须不断面对传染性微生物的变异和耐药菌株的发展,并研发新的药物来应对。因此,在这一领域的创新是必要的。迄今为止,已有三种肽类药物进入临床试验阶段,旨在治疗这些疾病。由于新冠病毒的大流行性扩散,去年所有抗感染肽类药物都进入了临床试验,以验证它们在治疗这种感染方面的可能用途。然而,由于这些重新定位研究并非基于针对蛋白质受体的药物化学方法,目前仍未获得可靠的结果。基于这些原因,我们决定将它们排除在本综述之外。
Nangibotide(LR12)是一种化学合成的12肽,源自TREM样转录物-1(TLT-1)的残基94至105。LR12由Inotrem公司开发,是一种特异性的TREM-1抑制剂,能够干扰TREM-1与其配体的结合。TREM-1通过与Toll样受体协同作用来放大先天性免疫反应,是脓毒性休克的关键介质。LR12通过与其配体结合来阻断TREM-1,并在脓毒症期间提供保护作用,如抑制过度反应、器官损伤和死亡,而不会造成有害后果。调节TREM-1信号传导的保护作用在其他炎症模型中也很明显,如胰腺炎、出血性休克和炎症性疾病。该药物目前正在进行2b期试验,用于治疗脓毒性休克,预计2021年下半年将公布结果。
Reltecimod(AB103,p2TA;CD288-15)是由 Atox Bio 发现的,作为一种通过减轻免疫失调来解决器官功能障碍或衰竭的候选药物。这是在坏死性软组织感染(NSTI)患者中经常出现的反应。它是一种合成类似物肽,与 T 淋巴细胞受体 CD28 的 8 - 15 片段同源。凭借其新颖的作用机制,它与共刺激受体的二聚体界面结合,并干扰 T 细胞上表达的 B7 - 2/CD28 的结合,从而调节导致全身器官衰竭的急性炎症。Reltecimod 已完成临床试验流程,目前正处于监管机构批准的过程中,这表明它将在不久的将来推向市场。
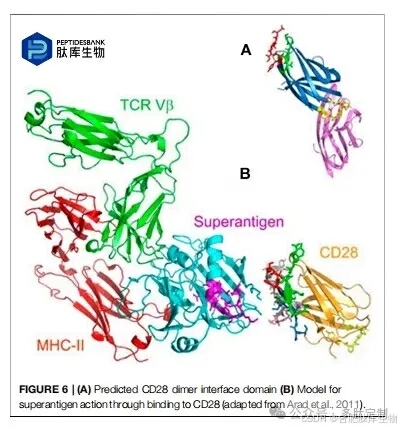
为了了解细菌超抗原毒素与 CD28 同聚体界面的结合,对 reltecimod(SPMLVAYD)进行了研究,它是通过与 CTLA-4 比对预测的 CD28 同聚体界面结构域的片段(图 6A)。在现有的 CD28 结构中,reltecimod 与同聚体界面重叠,参与了 CD28 中复杂的超抗原结合位点。该肽与细胞表面的 CD28 竞争超抗原中的β-链/铰链/α-螺旋结构域,位于远离其他蛋白质(MHC-II、TCR Vβ)结合位点的位点(图 6B)。阻断超抗原与 CD28 的相互作用足以阻止急性炎症、致命性中毒性休克。
C16G2 是由 Armata Pharmaceuticals 开发的,得益于一种用于治疗细菌感染的新型平台的设计,该平台被称为“特定靶向抗菌肽(STAMP)平台”。STAMPs 是通过挖掘细菌基因组以寻找赋予病原体特异性杀伤活性的靶向结构域而设计的。因此,C16G2 针对龋齿中的变形链球菌,是通过将一个靶向结构域(作为能力刺激肽 C16 信息素)与一个杀伤结构域相匹配而创建的,该靶向结构域能有效地在变形链球菌细胞表面积累。该领域,即 G2 序列,是广谱杀菌肽诺维司林 G 的截短版本。一个灵活的三甘氨酸序列将这两个区域连接起来。C16G2 已完成 2 期临床试验,表明该药物具有可接受的安全性和耐受性特征,并能选择性地减少口腔中的变形链球菌。然而,该公司决定不进行 3 期试验,以寻求降低 STAMP 商品成本的替代方案。
杂项
在本节中,收集了针对不同疾病的几种肽,因为它们无法归入本综述中所描述的特定病理领域之一。
LJPC-401 是一种合成的人类铁调素肽,由拉霍亚制药公司开发,用于潜在治疗以铁过载为特征的病症。在健康个体中,铁调素可防止重要器官(如肝脏和心脏)中铁的过度积累,因为铁的过度积累会导致严重的损害甚至死亡。
体内铁调素与铁水平之间的反馈回路确保了全身铁稳态。铁调素降低了细胞表面铁转运蛋白 - 1 受体(FPN - 1)的浓度,从而抑制了铁进入血浆。铁调蛋白在 N 和 C 结构域之间的中心腔与铁转运蛋白结合,起到分子帽的作用,阻碍铁外流通路。通过评估特定突变导致的结合亲和力降低,对肽与铁转运蛋白的极性和疏水相互作用网络进行了评估。
铁调素与 FPN-1 结合会导致受体发生改变,进而使其内化和在溶酶体中降解。通过这种方式,网状内皮巨噬细胞、肝细胞和十二指肠肠细胞的铁输出被阻断。铁调素缺乏是遗传性血色病(HH)的一个常见特征,LJPC-401 已完成针对这一治疗靶点的 2 期临床试验,这是一种“替代疗法”,用于补充不足的铁调素水平。
OXE-103(Oxeia Biopharmaceuticals)是一种合成的人类饥饿激素,是一种内源性激素,用于治疗脑震荡/轻度创伤性脑损伤。OXE-103 能够自由穿过血脑屏障,有助于稳定脑震荡后的代谢和能量功能障碍。OXE-103 独特地针对大脑的海马体区域,这是认知和记忆的重要区域。使用 OXE-103 治疗已被证明能够恢复正常的能量代谢,并减少在低能量状态下形成的活性氧物质的毒性作用。在神经元中,解偶联蛋白-2(UCP2)通过响应亚致死性应激来稳定线粒体。其确切的神经保护作用关于饥饿激素的作用仍不清楚,但有人提出丘脑神经元中依赖于 UCP2 的线粒体稳定作用。一项 2 期研究正在进行中,其目标是减轻 OXE103 治疗的症状负担。
索纳肽是一种合成的环状 17 肽,其分子结构模仿人类肿瘤坏死因子(TNF)的类凝集素结构域(TIP),与高原性肺水肿(HAPE)和成人呼吸窘迫综合征(ARDS)有关。索纳肽,也称为 AP-301,由 Apeptico Forschung und Entwicklung 公司开发,即将进入治疗各种肺部疾病的 2b 期。索纳肽通过直接与通道的关键α亚基结合并稳定其开放状态来激活肺上皮钠通道(ENaC),从而增强钠离子的摄取。TNF 的 TIP 结构域的寡糖结合特性在 TNF 和索纳肽与 ENaC 相互作用并激活其的机制中起着重要作用,尽管这种相互作用的确切性质尚不清楚。
TAK639(武田公司)是一种合成的9个氨基酸的线性肽,已完成治疗原发性开角型青光眼(POAG)和眼压(IOP)的1期临床试验。据估计,2020年全球有超过7500万人患有青光眼;特别是POAG是一种复杂的视神经病变,其特征是视神经和视网膜神经节细胞及其轴突的萎缩,导致不可逆的失明。临床证据表明,钠肽网络的表达和功能在包括人类小梁网在内的多个眼部系统中发挥关键作用。C型钠肽(CNP)是降低兔眼压最有效的钠肽,进一步研究表明,这些动物的眼中存在功能性的钠肽受体-B(NPR-B)。然而,这种有前景的治疗方法由于心房钠肽难以穿透角膜降低眼压,因此存在给药问题。为了克服这一缺陷,TAK639 被设计为一种可渗透角膜的 CNP 衍生物,其环鸟苷酸(cGMP)的产生和细胞浓度增加,能够降低眼压。
肽类作为治疗性药物的当前挑战与未来前景
工业药物发现和开发过程的细节通常在高度保密的情况下进行。大多数时候,结构会被尽可能长时间地保密,并且在产品专利中使用Markush 结构,不仅是为了扩大权利要求的范围,也是为了掩盖真正的候选药物配方。在我们追踪的 58 种肽中,有 11 种的结构未被披露。此外,对于异源肽,大多数时候,结合模式、蛋白质 - 蛋白质相互作用的热点、建模和 SARS 也会被保密,以尽可能推迟潜在的竞争对手,因为生成肽的“me-too”结构相当简单。
用于蛋白质-蛋白质相互作用的异源肽通常通过常规的小分子药物发现方法来鉴定,目标是主要通过肽库筛选来识别热点。一旦确定了命中点,结构变化的目标是将肽的长度尽可能缩短,以增加和稳定生物活性,结晶受体/配体或其片段,并明确识别产生对接模型的热点。候选药物的先导优化将进一步优化活性和选择性,并筛选可用于改善药理特征的氨基酸。
天然/类似肽的方法则完全不同,因为在这种情况下,活性肽序列通常已知,蛋白质-蛋白质相互作用也已知。关键问题在于建立特定的蛋白质-蛋白质相互作用与疾病之间的关系。生物信息学和对人类分子生理学的更好理解在减少实验方法中用于识别新靶点的时间和成本方面发挥着关键作用。从已经由自然界确定的天然序列开始,药物化学家的挑战与异源肽的挑战相似,目标是提交一份产品专利,在竞争激烈的环境中确定一系列新结构。
在天然/类似肽类药物开发方面,最显著的例子是针对 2 型糖尿病和肥胖症的 GLP-1R 激动剂。临床试验中的分子已在第五段中讨论,然而,就潜在市场和产业竞争而言,这是迄今为止最重要的 PPI,可用于关键评估。2 型糖尿病的首例 GLP-1R 激动剂艾塞那肽于 2012 年获得了美国食品药品监督管理局(FDA)的批准。该分子是艾塞那肽 - 4 的合成版本,艾塞那肽 - 4 是一种多肽,存在于吉拉怪物的唾液分泌中,其具有 53% 的与 GLP-1 的同源性。GLP-1 的半衰期为 1.5 - 5 分钟,艾塞那肽约为 2.4 小时。从那以后,另外五种与 GLP-1 或艾塞那肽具有极高同源性(> 90%)的分子已获批准,其中包括畅销药利拉鲁肽和司美格鲁肽。从图 7 中报告的比对结果可以看出,通过位点直接诱变和丙氨酸扫描确定的对于 GLP-1R 结合至关重要的氨基酸在所有肽段中均得以保留(残基 7、10、12、13、15、28、29、35、36)。
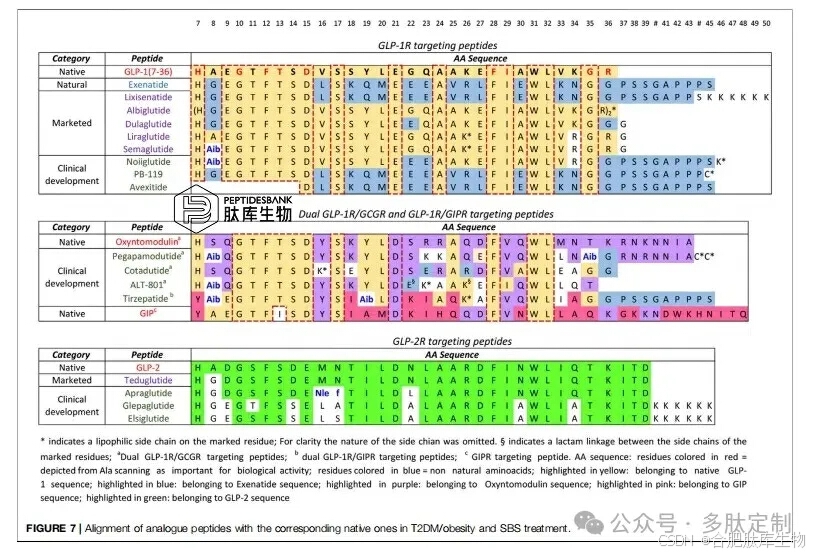
事实上,在 GLP-1 序列以及 GLP-1R/GIPR 和 GLP-1R/GCGR 双重抑制剂的结构中引入的所有修饰,其目的都是增加药理特征,同时不干扰结合效率。除了利拉鲁肽外,在所有已获批和临床试验的分子中,GLP-1 中对二肽基肽酶 - 4(DPP - 4)敏感的丙氨酸 8 已被甘氨酸、丙氨酸或丝氨酸所取代。此外,已知可延长半衰期并有可能提高口服生物利用率的侧链被引入其中六种分子中。临床试验中的七种分子已被附着到对 GLP-1R 结合并非关键的氨基酸上(残基 16、23、26、36、44、45 和 46)。从现有数据来看,司美格鲁肽在半衰期方面仍保持着纪录(7 天)。这些药物的总体开发情况已得到大量描述,临床试验中的新分子遵循了类似的开发路径。从科学角度来看,监测临床试验中双重和三重抑制剂的演变情况将很有趣,以了解这些方法相对于当前的畅销药是否会带来任何真正的治疗优势。
然而,偶然性也可能发挥重要作用,一个典型的例子是德鲁克博士发现 GLP-2 在肠道中的功能,最终促成了特度格鲁肽的商业化,用于治疗短肠综合征。天然 GLP-2 的半衰期非常短,只有 7 分钟,而在特度格鲁肽中,将丙氨酸在第二位替换为甘氨酸,使得该肽能够抵抗 DPP-4 的降解,从而实现了每日治疗所需的 2 小时半衰期,这与 GLP-1 类似物系列类似。换句话说,当了解 PPI 的生物学功能时,药物开发就会变得简单。
治疗性蛋白质(TP)的成功率始终高于小分子。肽类之所以具有与治疗性蛋白质类似的性能,主要是因为天然/类似物的作用以及它们高水平的选择性,这避免了非靶向毒性。另一方面,异源肽的成功率更类似于小分子,并且同样需要高水平的选择性来限制目标,将毒理学评估限制在免疫原性方面。当一个市场饱和时,管理和市场因素的影响开始对淘汰率产生重大影响。糖尿病和肥胖症吸引了多家公司的关注,因为从营业额的角度来看,这是迄今为止最重要的细分市场。我们已经描述了几种天然和类似物肽,它们针对 GLP-1R、GLP-1R/GCGR、GLP-1R/GIPR、Y2R,实际上已经在临床上使用了。然而,在这一领域已经有几种肽类药物获批,而且当新的药物获批时,几种强效且有效的药物将成为仿制药。因此,这些分子在临床上的成功率不仅将与疗效有关,还将与市场定位有关。
结论
正如整个肽类药物市场以及本文所讨论的大量在研项目所表明的那样,蛋白质 - 蛋白质相互作用(PPI)的调节是肽类药物研发的一个肥沃领域。本文所报道的概述表明,针对 PPI 是一种成功的途径,适用于许多情况。不同的病理情况,因为人类相互作用的网络调节着所有的生理级联事件及相关功能障碍。强大的合成方法论的可用性能够支持药物化学家在探索分子空间以设计具有改善药理性能的新结构方面。从技术角度来看,现在需要在合成技术的绿色化方面进行创新,以实现可持续性。
尽管进入临床试验途径的肽类药物数量一直在增加,但仍有大量蛋白质 - 蛋白质相互作用(PPI)尚未被探索或披露。事实上,重要的是要强调,除了与癌症相关的疾病以及糖尿病/肥胖症外,处于 1 期临床试验的分子数量非常有限。由于 PPI 可能产生的巨大影响,治疗性肽类药物的药物发现和开发过程以及监管和市场动态在不久的将来也会有所发展。总之,在理解 PPI 的背后,存在着蛋白质 - 蛋白质相互作用的圣杯,这将开启一个在安全有效药物的制药开发中肽类药物发挥重要作用的新时代。
免责声明:本文为行业交流学习,版权归原作者所有,如有侵权,可删除。


















 21
21

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









