






点击蓝字 关注我们
利用Parallel-Meta Suite在多平台下进行交互式微生物组分析

https://doi.org/10.1002/imt2.1

2022/3/6
● 2022年3月6日,青岛大学苏晓泉团队在iMeta在线发表题为“Parallel-Meta Suite: Interactive and rapid microbiome data analysis on multiple platforms”的研究性文章。开发了软件包Parallel-Meta Suite(PMS),可在多个平台上进行快速、全面的微生物组数据分析。
● 前文回顾▸▸▸ iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS(全文翻译,PPT,视频)
● 在该文的基础上,本文对PMS软件包
进行非常详细的逐步解读,方便读者使用。
● 第一作者:李坚
● 通讯作者:苏晓泉
(suxq@qdu.edu.cn)
摘 要
测序通量的提高和测序成本的降低,极大地方便了微生物组研究实验的开展,进而产生了浩如烟海的组测序数据,这些数据中蕴藏着微生物与其环境表型(如宿主健康或生态系统状态)之间的关联。想要破译隐藏在微生物组数据下的生物信息,出色而又可靠的软件工具是不可或缺的。然而现在的大多数的软件,其可用性方面的缺陷为非计算机专业的用户设置了难以逾越的鸿沟。与此同时,计算通量已经成为了许多分析平台处理大规模数据集的一个重要瓶颈。本研究开发了Parallel-Meta Suite(PMS),一个用于快速和全面的微生物组数据分析、可视化和注释的可交互软件套件。PMS采用了最先进的算法,涵盖序列微生物组数据物种与功能解析、统计分析、可视化等一系列流程,并具有友好的图形界面,可以满足各种用户的分析需求。为了适应快速增长的计算能力需求,PMS的整个分析流程都使用并行计算策略进行了优化,具备快速处理上万的样本的能力。此外,PMS还具有多操作系统兼容、简易安装与全自动运行等特性。
关键字:微生物组,宏基因组,扩增子,分析流程,可视化,并行计算
Bilibili:https://www.bilibili.com/video/BV16Y4y1v75k
Youtube:https://youtu.be/evrQXfL9ujE
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
仪器软件
● 仪器设备
目前Linux(如Ubuntu、CentOS、RedHat等)、Mac OS和Windows 10/11内置的WSL(Windows Subsystem for Linux)等操作系统均能够支持PMS。
PMS仅需要具有约2GB内存的标准计算机即可支持其安装与执行。为了更好的体验和更快的计算速度我们推荐在具有8GB以上内存和4核3.3Ghz以上CPU的标准计算机上使用PMS。
● 软件
PMS软件最新版本为3.7。该软件主要由C++和R语言开发编写。
C++语言需要安装C++编译器(例如g++)。对于Linux操作系统,大多版本已经在系统中安装了g++。对于Mac OS,建议从App Store安装Xcode应用程序,即可完成编译器的安装与配置。
R语言需要安装r-base提供运行环境。对于Linux操作系统,可以使用系统自带的包管理工具安装r-base。对于Mac OS,建议从App Store安装RStudio应用程序,即可完成R运行环境的安装与配置。
实验步骤
● 1. 安装Parallel-Meta Suite
我们建议选择步骤 1.1 中自动安装的方式来配置PMS软件。但如果自动安装程序失败,可以按照步骤 1.2 中的步骤手动安装PMS软件。
1.1 自动安装(首选方案)
1)下载对应操作系统的软件安装包
Linux和WSL的下载命令:
wget http://bioinfo.single-cell.cn/Released_Software/parallel-meta/3.7/parallel-meta-suite-3.7-src.tar.gzMacOS的下载命令:
curl -O http://bioinfo.single-cell.cn/Released_Software/parallel-meta/3.7/parallel-meta-suite-3.7-src-mac.tar.gz2)解压缩
使用以下命令对安装包进行解压缩:
tar –xzvf parallel-meta-suite-3.7-src.tar.gz3) 安装
运行以下安装命令:
cd parallel-meta-suite
source install.sh按照上述步骤操作,该软件包可以在30分钟内安装到计算机上,安装成功后提示信息如下(图1)所示:
图1. Parallel-Meta Suite安装成功提示信息
示例数据集在安装包内“examples”文件夹下,可以查看 “examples/Read me”中的内容来获取演示运行的详细信息,或直接运行:
sh Readme来自动演示示例数据集的处理运行。
该示例数据集包含一个文件夹和三个文件,其中,seqs文件夹中存放的是需要分析的样本序列,seqs.list为每个样本对应的序列存放路径。(格式详见表1),meta.txt为每个样本的meta信息(格式详见表2)。
1.2 手动安装(备选方案)
1)下载对应系统的安装包
与1.1步骤一致。
2)解压缩
与1.1步骤一致。
3)配置环境变量。
将以下内容,写入环境变量配置文件(Linux和Windows 10 WSL系统一般是$HOME/.bashrc,Mac系统一般是$HOME/.zshrc)。
export ParallelMETA=Path to Parallel-Meta Suite
export PATH="$PATH:$ParallelMETA/bin"
export PATH="$PATH:$ParallelMETA/Rscript"make并启用环境变量(如Linux下)
source ~/.bashrc4)安装R包
Rscript $ParallelMETA/Rscript/config.R5)编译源代码
cd parallel-meta-suite
make● 2. 输入格式
2.1 样本序列
一个序列文件中包含单个样本的所有测序数据。PMS可以接受fastq和fasta格式的测序数据。序列可以是扩增子测序序列(包括16S rRNA gene、18S rRNA gene和ITS gene),也可以是宏基因组鸟枪法测序序列(shotgun metagenome)。
2.2 样本列表
样本列表为纯文本格式文件,其中含有多个样本的ID和测序数据文件的地址路径(表1)。该文件有两列信息,第一列为样本的ID,第二列表示每个样本测序数据文件的路径。为了保证路径的合法性,我们强烈建议使用绝对地址(即包含完整的路径名称,如表1所示)。

表1. 文件列表格式
2.3 Meta信息
Meta信息文件为纯文本格式文件,包含测序样本的meta信息,通常会有多列,其中第一列是样本的ID,其他列为meta信息的项目,如表2所示。需要注意的是,样本ID命名及其顺序需要与样本列表中的样本ID保持一致。

表2.meta信息文件格式
● 3. 分析
3.1 自动化分析流程
PMS具有一个图形化交互式的“配置向导”,位于程序包中的PMS-config文件夹中,其名称为“index.html”。用网页浏览器打开后可以看到其主界面,如图2所示。在初始状态下,所有的参数已设为默认值,只需填入必要的基本参数(如输入/输出类型和路径)就可以进行分析。也可以调整高级选项,以进一步对剖析、多样性分析和统计这些步骤进行定制。最后,根据用户的设置,该配置向导可以生成相应的可执行命令。
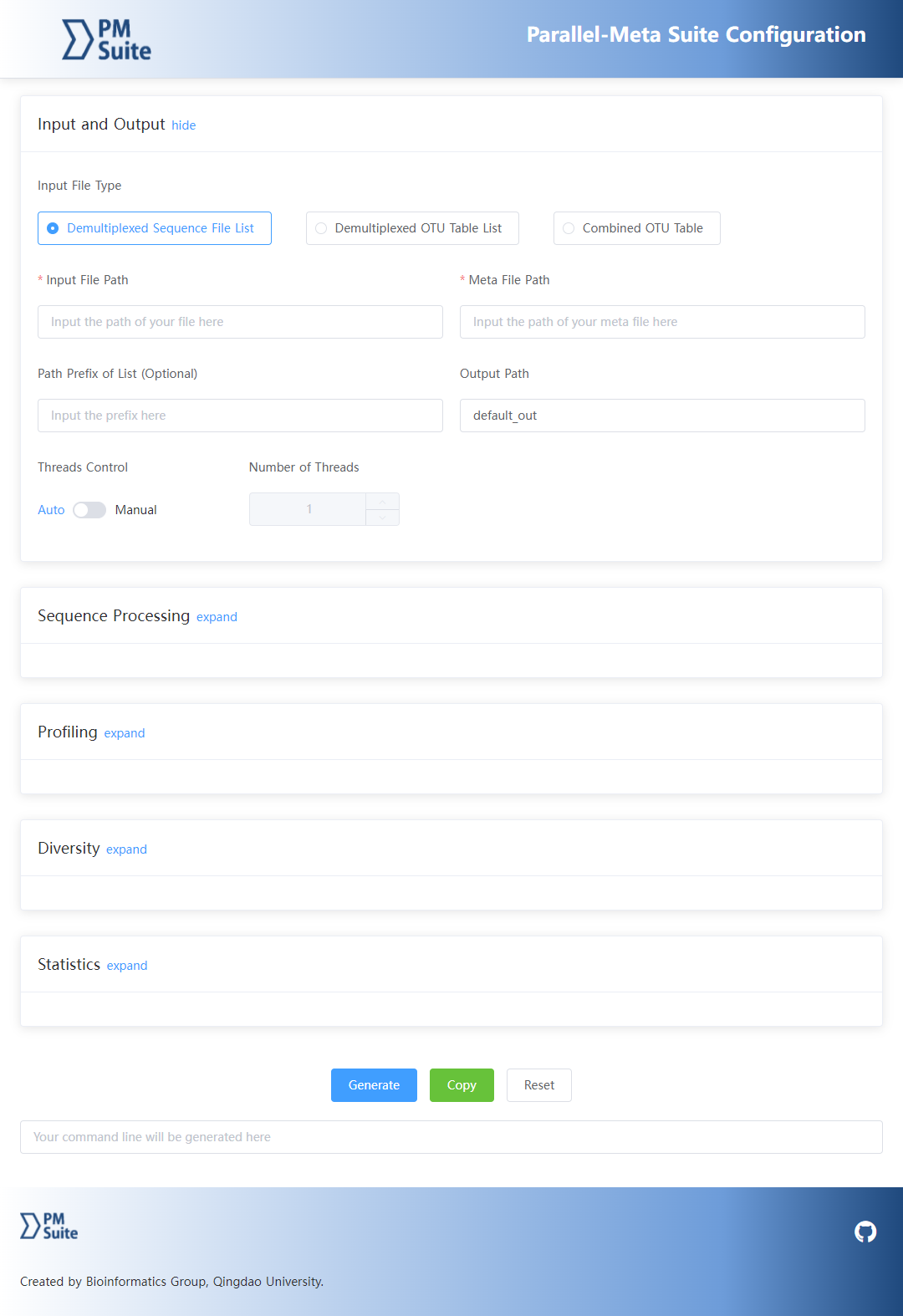
图2. 配置向导页面
在此,我们将展示PMS在不同计算平台和环境下的三个典型场景的使用情况和经验(图3)。需注意的是配置指南是可以独立运行的,配置过程可和分析过程可能在不同设备和环节下执行,但输入输出文件路径还是要以分析执行的设备为准。
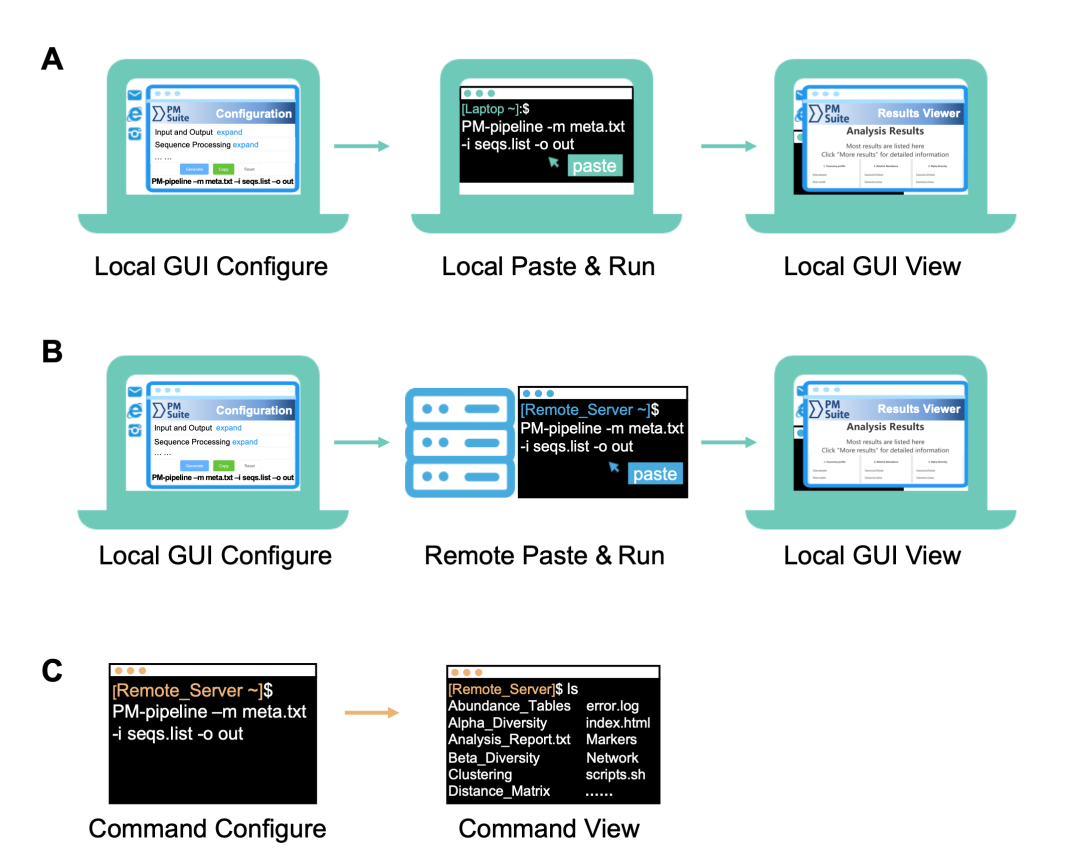
图3. PMS在不同场景和平台的三种典型使用方式
(A)在本机使用配置向导进行参数配置,并在本机进行运算分析;(B)在本机使用配置向导进行参数配置,并在远程服务器上进行运算分析;(C)使用命令行进行参数配置(本地和远程均可)
3.1.1 场景一:在本地使用配置向导进行参数配置,并在本地进行运算分析
PMS可以在“本地”个人电脑(如笔记本电脑)中安装和执行,以处理少量样品(比如少于200;图3A)。该场景适用于Linux(安装GUI桌面)、Mac或Windows 10+(需要安装Windows Subsystems for Linux(WSL))操作系统。配置完成后,通过点击页面底部的“Generate”和“Copy”按钮,就会生成一条有效的命令并复制到剪贴板中。然后将这个单行命令粘贴在本地终端,就可以成功运行PMS分析流程,而不需要进行其他操作。
3.1.2 场景二:在本地使用配置向导进行参数配置,并在远程服务器上进行运算分析
大量样本(比如大于1,000)的处理和运算需要更长的时间和更多的计算资源,我们建议在更强大的服务器上运行PMS的分析流程。通常这样的服务器需要远程登录(例如,通过SSH),并且只提供一个基于命令的终端来操作软件。在这种情况下(图3B),用户应在服务器上安装PMS,在本地计算机下载并打开配置向导(下载软件包中PMS-config文件夹,并用浏览器打开其中的“index.html”文件)以生成命令,并在远程服务器的终端上运行这些命令。因此,整个分析流程可以很容易地配置和执行,而无需大量的数据传输。
3.1.3 场景三:使用命令行进行参数配置
PMS也支持基于命令行的操作,此种方式通常是在没有GUI的条件下,或者针对有经验的用户(图3C)。整个分析流程可以在高度灵活的配置下工作,例如,用定制的参数运行每个步骤,或者只执行工作流程中的选定步骤。命令行界面还提供了教程,描述了详细的用法和分析流程在每个单一步骤中的简要帮助信息,可以通过“-h”参数来查看。以下命令是基于命令行操作中的简单范例:
PM-pipeline -i seqs.list -m meta.txt -o output其中,PM-pipeline是PMS的自动化分析程序;“seqs.list”是输入的样本扩增子测序序列列表(表1),用“-i”指定;“meta.txt”是输入meta信息(表2),用“-m”指定;“output”是输出文件夹,用“-o”指定。
3.2 查看结果
整个分析流程完成后,会在输出目录中自动创建结果导览,其文件名为“index.html”,可用网页浏览器打开。该页面会将所有分析结果分类(图4),为微生物组分析结果提供直接和清晰的解释。此外,在输出目录中,所有的原始结果(如相对丰度表、距离矩阵等)也会保留(表3),用于进一步深入的数据挖掘或元分析。此外,在结果文件夹中还提供了分析总结、工作日志和详细的分步工作流程脚本。

图4. PMS的结果导览页面

表3. 输出目录的文件列表
3.3 中间结果重分析
除了测序序列之外,PMS还可以接受以中间结果作为输入,对其进行重分析,例如,以样本丰度表或中间结果列表作为输入(表3),从而避免了重复的序列处理所造成的计算时间和资源的消耗。在图2的配置向导中,选择输入类型为“Demultiplexed OTU Table List”,可将中间结果列表(例如,输出目录中“Single_Sample.List”文件夹下的“taxa.list”文件)作为输入;或者选择输入类型为“Combined OTU Table”,可将样本丰度表(例如,输出目录中“Abundance_Tables”文件夹下的“taxa.OTU.Count”文件)作为输入。此外,命令行操作中也有相应的输入格式,例如:
PM-pipeline -l output/Single_Sample.List/taxa.list -m meta.txt -o output_new
## 或者
PM-pipeline -T output/Abundance_Tables/taxa.OTU.Count -m meta.txt -o output_new即为以上配置向导中等效的配置命令。
● 4. Parallel-Meta Suite的工作流程
PMS的分析工作流程如图5所示。PMS可以接受宏基因组的鸟枪序列或扩增子序列作为原始输入。对于鸟枪法测序序列,利用隐式马尔可夫模型(Mistry等, 2013)识别和提取标记基因片段(如16S rRNA或18S rRNA基因)。对于扩增子序列,PMS对标记基因进行ASV降噪(Callahan等, 2017)和去嵌合体(Edgar等, 2011),以降低测序错误的干扰(这一步骤对于鸟枪法测序序列的默认设置是关闭,也可由用户自行开启)。然后,通过内置的vsearch(Rognes等, 2016)将序列与参考数据库进行比对,进行从界级到物种级的剖析和分类学注释。每个分类级别上群落成员的相对丰度也使用标记基因拷贝数进行校正。之后,使用PICRUSt算法(Douglas等, 2020)预测功能信息的KEGG Orthology(KO)基因家族,并通过KEGG BRITE层次结构对代谢途径进行注释。PMS还通过NSTI(Nearest Sequenced Taxonomy Index)值来衡量功能的预测准确性(Langille等, 2013),NSTI是由OTU和它们在系统发育结构中最近的单独测序的亲属之间的距离之和计算出来。
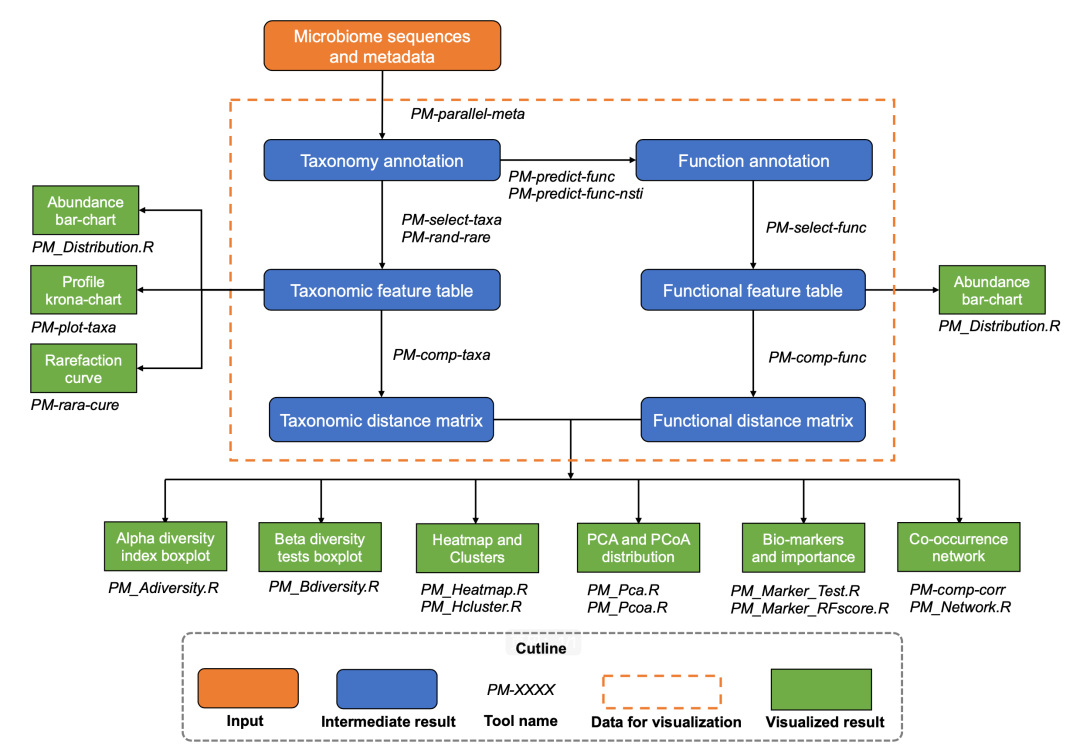
图5. PMS的工作流程
微生物组的物种信息通过Krona(Ondov等, 2011)和条形图进行可视化。然后,在用户选择的特定分类学或路径级别上进行微生物多样性分析、生物标记物选择和共现网络构建。α多样性分析计算每个样品的香农、辛普森和Chao1指数。对于离散的元数据(如类型、状态、性别等),α多样性指数进行Wilcoxon或Kruskal秩和检验,对于连续变量(如年龄、BMI、PH值等)进行回归分析。β多样性通过加权/非加权Meta-Storms(Su等, 2012)算法(针对物种分类)或Hierarchical Meta-Storms(Zhang等, 2021)(针对功能)计算所有样本之间距离矩阵,并通过热图进行可视化。之后,通过PCoA(主坐标分析)和PCA(主成分分析)图展示β-多样性模式,对离散元数据进行PERMANOVA和ANOSIM检验,对连续变量和距离值进行回归分析。在生物标志物分析中,PMS使用Wilcoxon或Kruskal秩和检验,选择出在不同组别(离散数据变量)间具有显著差异的微生物或基因单元作为候选标记物,然后通过随机森林(Vangay等, 2019; Qian等, 2020)的重要性进行排序。与连续变量密切相关的微生物组特征也通过回归分析被挑选出来作为生物标志物。在共现网络中,网络节点是群落特征(例如,一个微生物分类单元),网络的边代表节点间的Spearman相关性,然后计算网络密度、直径、半径和集中度来量化网络属性。
结果与分析
为了证明PMS在解码微生物组概况和将生态模式与关键meta数据联系起来的能力,这里选取了医院开业前后室内微生物组的变化验证。所有的数据集均可在 PMS 软件下载页面的“Supplementary”部分中下载。
数据集包含894个来自医院开业前后室内环境的16S-扩增子微生物组样本。我们用所有的默认参数执行了PMS分析流程。从结果中我们可以观察到,医院开放后,α多样性的香农指数下降(图6A;Wilcoxon检验p值<0.01),整体群落的β多样性明显转变(图6B;加权Meta-Storms距离,PERMANOVA检验p值<0.01),均已被Lax等人(Lax等, 2017)验证过。两个时间点之间的这种微生物动态也可以通过相对丰度的变化来说明(图6C)。使用统计测试和机器学习分析方法,PMS还确定了有助于区分医院表面从开业前到开业后状态的这种生态变化的最重要的微生物,如葡萄球菌、莱茵海拉菌和莫德斯特菌。这个机器学习模型在区分室内样本(图6D)的属级状态方面达到了95.91%的准确率(误差率=4.09%)。
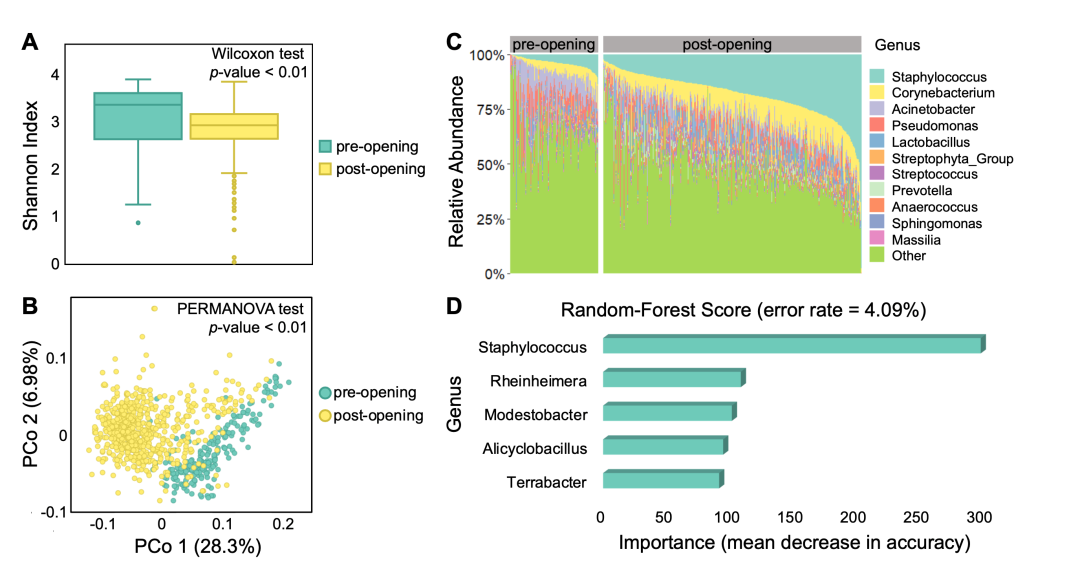
图6. 医院开业前后室内微生物组的变化
(A)医院开业后,α多样性的香农指数下降,Wilcoxon测试P值<0.01(P值<0.05表示差异显著);
(B)根据加权的Meta-Storms距离,开院前和开院后状态下的整体β多样性有显著差异,PERMANOVA检验P值<0.01;
(C)两个时间点之间属水平的相对丰度的动态变化;
(D)五种细菌属被选为可以区分两个时间点的生物标志物。X轴是随机森林模型产生的重要性得分(准确性的平均下降),该模型评估了每个生物标志物对区分不同医院状态的重要性
失败经验
● 问题1
安装提示:“make: g++: command not found”
问题原因:没有安装Parallel-Meta Suite所需要的g++编译器。
解决方法:根据不同的操作系统,利用相应的命令安装 g++,常见的操作系统:
Ubuntu Linux系统:sudo apt-get install g++
CentOS Linux系统:sudo yum install g++
Mac OS 系统:通过App Store安装Xcode应用程序
● 问题2
运行提示:“Please set the environment variable ParallelMETA to the directory”
问题原因:环境变量设置失败。
解决方法:请参考实验步骤 1.2.2 中手动配置环境变量的方法将 Parallel-Meta Suite 所需要的环境变量添加到配置文件中。
● 问题3
运行提示:“PM-pipeline: command not found”
问题原因:环境变量设置失败。
解决方法:请参考实验步骤 1.2.2 中手动配置环境变量的方法将 Parallel-Meta Suite 所需要的环境变量添加到配置文件中。
● 问题4
运行提示:“Error: Cannot open file: XXX”
问题原因:输入了错误的输入/输出文件路径。
解决方案:请检查正确的输入文件路径(可在输入时用Tab 键自动补全),并确保用户在输出路径下有足够的写权限。
● 问题5
运行提示:“Argument #X Error : Arguments must start with -”
问题原因:运行命令中所有参数选项名称必须以“-”开头。
解决方法:请检查第 X 个参数并更正。
致谢
本项工作得到了国家重点研发计划2021YFF0704500、国家自然科学基金31771463和32070086项目的支持。
引文格式
Yuzhu Chen, Jian Li, Yufeng Zhang, Mingqian Zhang, Zheng Sun, Gongchao Jing, Shi Huang, Xiaoquan Su. 2022. Parallel-Meta Suite: Interactive and rapid microbiome data analysis on multiple platforms. iMeta 1: e1. https://doi.org/10.1002/imt2.1
作者简介

陈俞竹
● 青岛大学软件工程学术硕士,2019年公派至瑞典布莱津理工大学交换学习。
● 目前研究方向为微生物组大数据分析与挖掘,相关学术成果已发表于iMeta、Computational and Structural Biotechnology Journal等期刊。

李坚
● 青岛大学电子信息专业硕士。
● 前中兴通讯工程师,后考入青岛大学攻读硕士学位。目前研究的主要课题为微生物组分析工具。

苏晓泉(通讯作者)
● 青岛大学教授,博士生导师。
● 研究方向为生物信息学与大数据科学,已在mBio、mSystems、Bioinformatics、iMeta等期刊发表学术论文40余篇,主持国家自然科学基金项目、国家重点研发子课题、山东省自然基金重大基础项目、中科院重点部署项目子课题等,相关成果获得8项软件著作权。
更多推荐
(▼ 点击跳转)
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
iMeta:哈佛刘洋彧等基于物种组合预测菌群结构的深度学习方法
iMeta:吴青龙/王明福/刘金鑫等-从肠道菌群看待人类对高原饮食的适应性
iMeta:西农韦革宏团队焦硕等-土壤真菌驱动细菌群落的构建
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

微信公众号
iMeta
责任编辑
微微
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
















 6305
6305











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









