






DeepST共三项工作,聚类分析、单细胞和空间转录组数据联合分析(解卷积)、空间对齐。第一项任务已经看完代码了,现在看第二项联合分析。
此项以人类淋巴结Human_Lymph_Node数据为例(10X),作者已经处理好数据,并上传到谷歌云盘上了,直接下载用就好了。
原始数据如下。
https://support.10xgenomics.com/spatial-gene-expression/datasets/1.0.0/V1_Human_Lymph_Node
下面还是看代码。
import scanpy as sc
from DeepST import DeepST
dataset = 'Human_Lymph_Node'
file_path = 'Human_Lymph_Node/'
# set random seed
random_seed = 50
DeepST.fix_seed(random_seed)
# read ST data
adata = sc.read_h5ad(file_path+'ST.h5ad')
adata.var_names_make_unique()
# preprocessing for ST data
DeepST.preprocess(adata)
# build graph
DeepST.construct_interaction(adata)
DeepST.add_contrastive_label(adata)
# read scRNA daa
adata_sc = sc.read(file_path+'scRNA.h5ad')
adata_sc.var_names_make_unique()
# preprocessing for scRNA data
DeepST.preprocess(adata_sc)以上主函数,直到这里,都没有新的代码,具体可看上一篇博客。具体包括数据预处理,构建正负类标签、邻接矩阵等。
然后挑选单细胞数据和空间转录组数据的共同基因。
def filter_with_overlap_gene(adata, adata_sc):
# remove all-zero-valued genes
#sc.pp.filter_genes(adata, min_cells=1)
#sc.pp.filter_genes(adata_sc, min_cells=1)
if 'highly_variable' not in adata.var.keys():
raise ValueError("'highly_variable' are not existed in adata!")
else:
adata = adata[:, adata.var['highly_variable']]
if 'highly_variable' not in adata_sc.var.keys():
raise ValueError("'highly_variable' are not existed in adata_sc!")
else:
adata_sc = adata_sc[:, adata_sc.var['highly_variable']]
# Refine `marker_genes` so that they are shared by both adatas
genes = list(set(adata.var.index) & set(adata_sc.var.index))
genes.sort()
print('Number of overlap genes:', len(genes))
adata.uns["overlap_genes"] = genes
adata_sc.uns["overlap_genes"] = genes
adata = adata[:, genes]
adata_sc = adata_sc[:, genes]
return adata, adata_sc分别筛选单细胞核空转的高表达基因,然后挑选共同的基因。
继续,构建正负类特征,这个也已介绍。
DeepST.get_feature(adata)继而,构建模型。
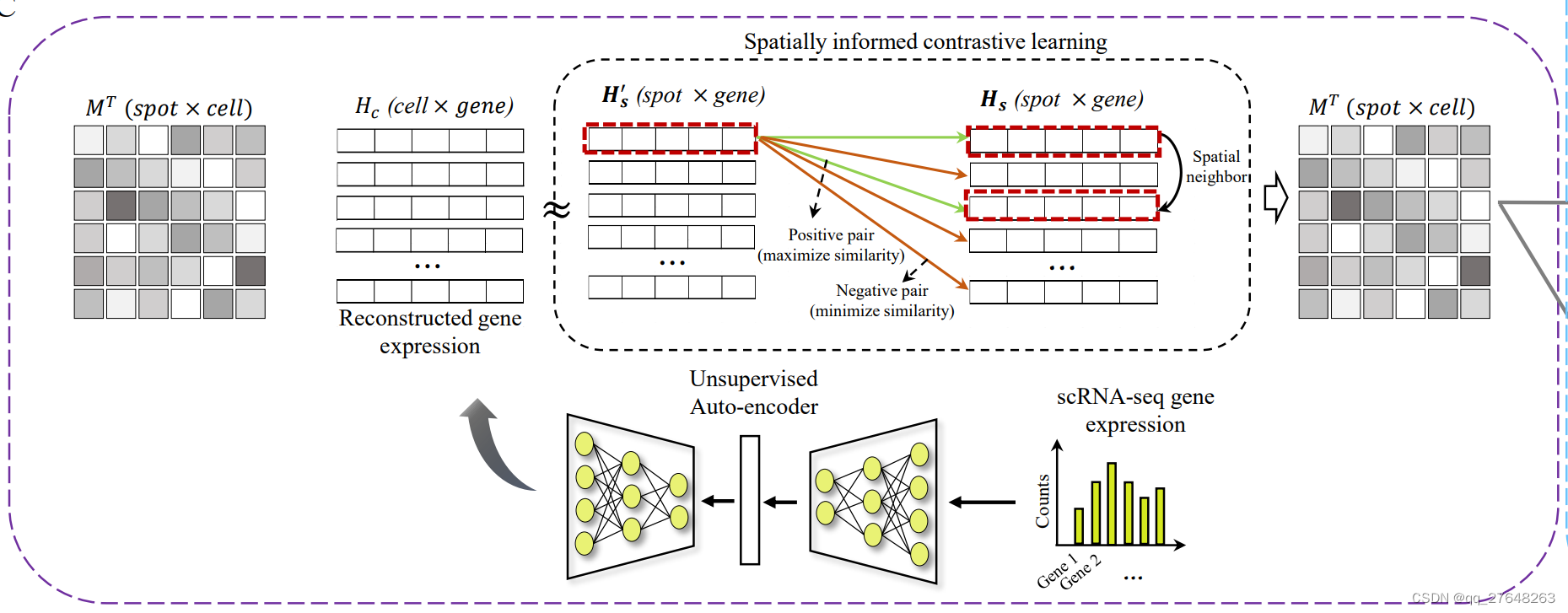
model = DeepST.Train(adata, adata_sc, epochs=1200, deconvolution=True)这里解卷积就是为真了,并且epochs变大了,模型结构与之前一致,这里就不赘述了。
初始化程序不放上了,用到的时候会再说。
判断解卷积,在缺失值位置10X数据正负类赋值为0.
if self.deconvolution:
self.adata_sc = adata_sc.copy()
if isinstance(self.adata.X, csc_matrix) or isinstance(self.adata.X, csr_matrix):
self.feat_sp = adata.X.toarray()[:, ]
else:
self.feat_sp = adata.X[:, ]
if isinstance(self.adata_sc.X, csc_matrix) or isinstance(self.adata_sc.X, csr_matrix):
self.feat_sc = self.adata_sc.X.toarray()[:, ]
else:
self.feat_sc = self.adata_sc.X[:, ]
# fill nan as 0
self.feat_sc = pd.DataFrame(self.feat_sc).fillna(0).values
self.feat_sp = pd.DataFrame(self.feat_sp).fillna(0).values
self.feat_sc = torch.FloatTensor(self.feat_sc).to(self.device)
self.feat_sp = torch.FloatTensor(self.feat_sp).to(self.device)
if self.adata_sc is not None:
self.dim_input = self.feat_sc.shape[1]
self.n_cell = adata_sc.n_obs
self.n_spot = adata.n_obs仔细看下模型训练,这个和之前有不少不同的。
adata, adata_sc = model.train_map()首先是训练空间转录组的模型,然后提取特征,这个和之前的没有任何区别,不做介绍了。
emb_sp = self.train()然后训练单细胞数据的模型
emb_sc = self.train_sc()首先构建编码器,这里选取的是六层线性层编码器(三层编码,三层解码),输出是重构的单细胞数据(输入输出维度相同,这个和空转是一样的)。
class Encoder_sc(torch.nn.Module):
def __init__(self, dim_input, dim_output, dropout=0.0, act=F.relu):
super(Encoder_sc, self).__init__()
self.dim_input = dim_input
self.dim1 = 256
self.dim2 = 64
self.dim3 = 32
self.act = act
self.dropout = dropout
#self.linear1 = torch.nn.Linear(self.dim_input, self.dim_output)
#self.linear2 = torch.nn.Linear(self.dim_output, self.dim_input)
#self.weight1_en = Parameter(torch.FloatTensor(self.dim_input, self.dim_output))
#self.weight1_de = Parameter(torch.FloatTensor(self.dim_output, self.dim_input))
self.weight1_en = Parameter(torch.FloatTensor(self.dim_input, self.dim1))
self.weight2_en = Parameter(torch.FloatTensor(self.dim1, self.dim2))
self.weight3_en = Parameter(torch.FloatTensor(self.dim2, self.dim3))
self.weight1_de = Parameter(torch.FloatTensor(self.dim3, self.dim2))
self.weight2_de = Parameter(torch.FloatTensor(self.dim2, self.dim1))
self.weight3_de = Parameter(torch.FloatTensor(self.dim1, self.dim_input))
self.reset_parameters()
def reset_parameters(self):
torch.nn.init.xavier_uniform_(self.weight1_en)
torch.nn.init.xavier_uniform_(self.weight1_de)
torch.nn.init.xavier_uniform_(self.weight2_en)
torch.nn.init.xavier_uniform_(self.weight2_de)
torch.nn.init.xavier_uniform_(self.weight3_en)
torch.nn.init.xavier_uniform_(self.weight3_de)
def forward(self, x):
x = F.dropout(x, self.dropout, self.training)
#x = self.linear1(x)
#x = self.linear2(x)
#x = torch.mm(x, self.weight1_en)
#x = torch.mm(x, self.weight1_de)
x = torch.mm(x, self.weight1_en)
x = torch.mm(x, self.weight2_en)
x = torch.mm(x, self.weight3_en)
x = torch.mm(x, self.weight1_de)
x = torch.mm(x, self.weight2_de)
x = torch.mm(x, self.weight3_de)
return x然后是训练模型,损失函数就是输入和输出的MSE。
def train_sc(self):
self.model_sc = Encoder_sc(self.dim_input, self.dim_output).to(self.device)
self.optimizer_sc = torch.optim.Adam(self.model_sc.parameters(), lr=self.learning_rate_sc)
print('Begin to train scRNA data...')
for epoch in tqdm(range(self.epochs)):
self.model_sc.train()
emb = self.model_sc(self.feat_sc)
loss = F.mse_loss(emb, self.feat_sc)
self.optimizer_sc.zero_grad()
loss.backward()
self.optimizer_sc.step()
print("Optimization finished for cell representation learning!")
with torch.no_grad():
self.model_sc.eval()
emb_sc = self.model_sc(self.feat_sc)
return emb_sc随后将单细胞数据和空转数据数据正则化一下,构建映射模型。
self.adata.obsm['emb_sp'] = emb_sp.detach().cpu().numpy()
self.adata_sc.obsm['emb_sc'] = emb_sc.detach().cpu().numpy()
# Normalize features for consistence between ST and scRNA-seq
emb_sp = F.normalize(emb_sp, p=2, eps=1e-12, dim=1)
emb_sc = F.normalize(emb_sc, p=2, eps=1e-12, dim=1)
self.model_map = Encoder_map(self.n_cell, self.n_spot).to(self.device)
self.optimizer_map = torch.optim.Adam(self.model_map.parameters(), lr=self.learning_rate, weight_decay=self.weight_decay)
再来看Encoder_map模型结构,传入的参数分别是单细胞数据细胞个数和空转数据spot个数(以这两个参数为行和列构建了一个矩阵为映射矩阵,最终也是要训练得到映射矩阵)。
class Encoder_map(torch.nn.Module):
def __init__(self, n_cell, n_spot):
super(Encoder_map, self).__init__()
self.n_cell = n_cell
self.n_spot = n_spot
self.M = Parameter(torch.FloatTensor(self.n_cell, self.n_spot))
self.reset_parameters()
def reset_parameters(self):
torch.nn.init.xavier_uniform_(self.M)
def forward(self):
x = self.M
return x 再来看映射训练,生成M矩阵,计算损失函数,优化参数。
for epoch in tqdm(range(self.epochs)):
self.model_map.train()
self.map_matrix = self.model_map()
loss_recon, loss_NCE = self.loss(emb_sp, emb_sc)
loss = self.lamda1*loss_recon + self.lamda2*loss_NCE
self.optimizer_map.zero_grad()
loss.backward()
self.optimizer_map.step()具体来看损失函数。损失函数包括两部分,重构损失和对比损失(不太确定这么形容对不对)。首先由M经过softmax后得到map_probs,转置后与单细胞编码数据相乘即为重构空间转录组数据。
def loss(self, emb_sp, emb_sc):
'''\
Calculate loss
Parameters
----------
emb_sp : torch tensor
Spatial spot representation matrix.
emb_sc : torch tensor
scRNA cell representation matrix.
Returns
-------
Loss values.
'''
# cell-to-spot
map_probs = F.softmax(self.map_matrix, dim=1) # dim=0: normalization by cell
self.pred_sp = torch.matmul(map_probs.t(), emb_sc)
loss_recon = F.mse_loss(self.pred_sp, emb_sp, reduction='mean')
loss_NCE = self.Noise_Cross_Entropy(self.pred_sp, emb_sp)
return loss_recon, loss_NCE重构的空间转录组数据与原始的空间转录组编码数据的MSE值即为重构损失。对比损失这一部分可以参照SimCLR模型,即相近的越相似越好,不相近的越不相似越好,这里的相近指的空间相近,相似指的余弦相似度。具体来说,每一个spot和其邻接矩阵中有连接的spot为正类,其他spot为负类,这个方法最初在陈洛南的stMVC中图像特征提取中见到过。
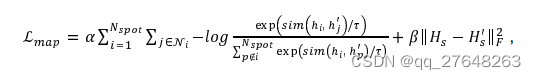
具体来看下对比损失函数,余弦相似度的程序不放进去了。
计算分母的时候(k),是不是应该是
k=torch.exp(mat-torch.diag_embed(torch.diag(a, 0))).sum(axis=1)
其他的都是按照公式计算的,请路过大佬指教。
def Noise_Cross_Entropy(self, pred_sp, emb_sp):
'''\
Calculate noise cross entropy. Considering spatial neighbors as positive pairs for each spot
Parameters
----------
pred_sp : torch tensor
Predicted spatial gene expression matrix.
emb_sp : torch tensor
Reconstructed spatial gene expression matrix.
Returns
-------
loss : float
Loss value.
'''
mat = self.cosine_similarity(pred_sp, emb_sp)
k = torch.exp(mat).sum(axis=1) - torch.exp(torch.diag(mat, 0))
# positive pairs
p = torch.exp(mat)
p = torch.mul(p, self.graph_neigh).sum(axis=1)
ave = torch.div(p, k)
loss = - torch.log(ave).mean()
return loss这样,经过三次训练,就得到了空转编码数据,单细胞编码数据,映射矩阵。
with torch.no_grad():
self.model_map.eval()
emb_sp = emb_sp.cpu().numpy()
emb_sc = emb_sc.cpu().numpy()
map_matrix = F.softmax(self.map_matrix, dim=1).cpu().numpy() # dim=1: normalization by cell
self.adata.obsm['emb_sp'] = emb_sp
self.adata_sc.obsm['emb_sc'] = emb_sc
self.adata.obsm['map_matrix'] = map_matrix.T # spot x cell
return self.adata, self.adata_sc继续,做单细胞和空转数据映射。
from DeepST.utils import project_cell_to_spot
project_cell_to_spot(adata, adata_sc, retain_percent=0.15)具体来看程序。
1.只保留映射矩阵前百分之十的值,其他值置零。
2.根据单细胞数据的标签,构建单细胞类型矩阵。
3.映射矩阵乘单细胞标签矩阵即空转标签矩阵。
4.空转标签矩阵正则化。
def project_cell_to_spot(adata, adata_sc, retain_percent=0.1):
'''\
Project cell types onto ST data using mapped matrix in adata.obsm
Parameters
----------
adata : anndata
AnnData object of spatial data.
adata_sc : anndata
AnnData object of scRNA-seq reference data.
retrain_percent: float
The percentage of cells to retain. The default is 0.1.
Returns
-------
None.
'''
# read map matrix
map_matrix = adata.obsm['map_matrix'] # spot x cell
# extract top-k values for each spot
map_matrix = extract_top_value(map_matrix,retain_percent) # filtering by spot
# construct cell type matrix
matrix_cell_type = construct_cell_type_matrix(adata_sc)
matrix_cell_type = matrix_cell_type.values
# projection by spot-level
matrix_projection = map_matrix.dot(matrix_cell_type)
# rename cell types
cell_type = list(adata_sc.obs['cell_type'].unique())
cell_type = [str(s) for s in cell_type]
cell_type.sort()
#cell_type = [s.replace(' ', '_') for s in cell_type]
df_projection = pd.DataFrame(matrix_projection, index=adata.obs_names, columns=cell_type) # spot x cell type
#normalize by row (spot)
df_projection = df_projection.div(df_projection.sum(axis=1), axis=0).fillna(0)
#add projection results to adata
adata.obs[df_projection.columns] = df_projection最后进行可视化。
# Visualization of spatial distribution of scRNA-seq data
import matplotlib as mpl
with mpl.rc_context({'axes.facecolor': 'black',
'figure.figsize': [4.5, 5]}):
sc.pl.spatial(adata, cmap='magma',
# selected cell types
color=['B_Cycling', 'B_GC_LZ', 'B_GC_DZ', 'B_GC_prePB'],
ncols=4, size=1.3,
img_key='hires',
# limit color scale at 99.2% quantile of cell abundance
vmin=0, vmax='p99.2'
)















 589
589











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









