







目录
2.2 数据对齐(pairwise slice alignment)
2.3 数据整合(center slice integration)
0 参考文献
PASTE原论文、补充说明、代码等:Alignment and integration of spatial transcriptomics data
1 背景介绍
空间转录组学(Spatial Transcriptomics)是一种生物学和生物信息学领域的技术和方法,它用于研究组织、细胞和生物样本中基因表达的空间分布。这个领域的目标是理解基因在生物组织中的位置和表达模式,以帮助解释组织发育、疾病机制和其他生物学过程。
传统的转录组学研究主要关注基因的表达水平(mRNA expression / gene expression),但没有提供关于基因表达在组织中的空间分布的信息。空间转录组学通过将组织中的基因表达数据与其空间位置相关联,使研究人员能够获得有关哪些基因在特定区域或细胞类型中表达得更多或更少的信息。这种技术通常包括高通量测序和图像分析,以获取组织切片的分子信息。
现有的可以从组织切片同时获取基因表达水平和位置信息的技术如10x Visium的原理,可以参考我的另一篇博客【技术介绍】空间转录组学10X Visium方法原理
2 算法解析
尽管许多ST研究从多个相邻的组织切片中生成数据,但几乎所有当前的ST分析技术要么分析单个切片,要么在不考虑空间坐标的情况下跨切片汇集基因表达数据。然而,单个切片的分析对于检测跨空间变化的低表达转录本的能力较低。
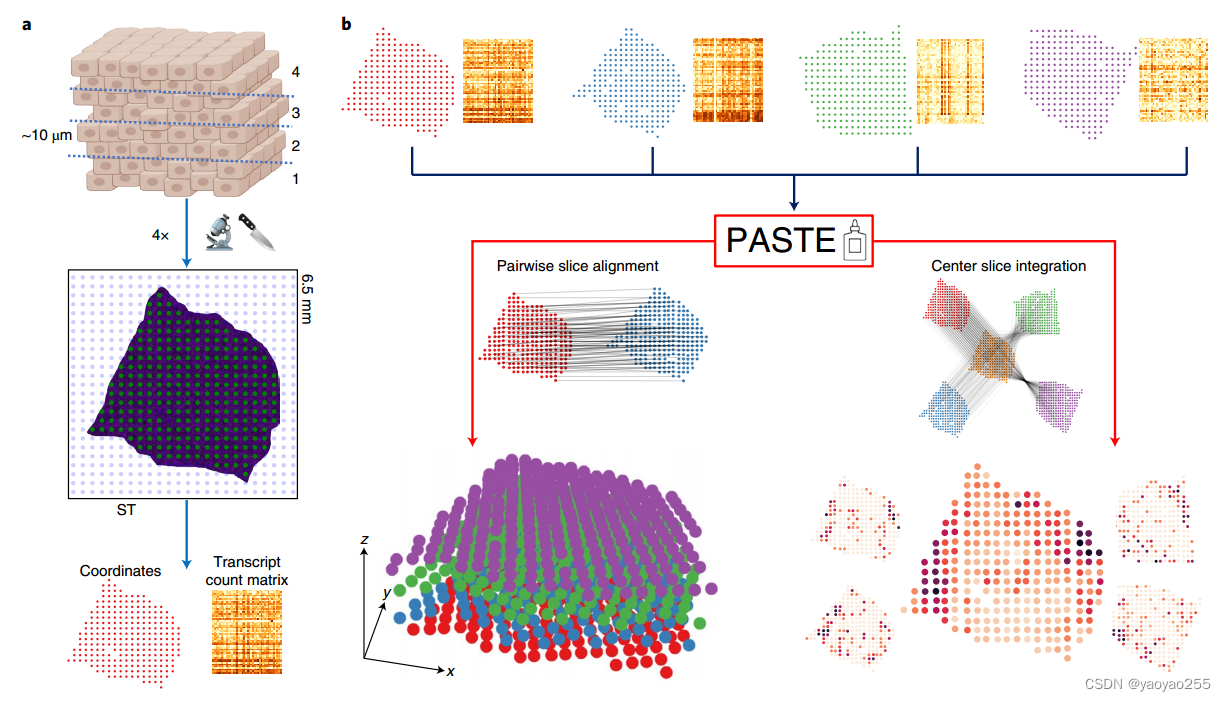
PASTE(probabilistic alignment of Spacial Transcriptomics experiments)是一种通过同时利用数据中提供的基因表达水平信息和位置信息,将相邻组织切片通过所获得的数据进行对齐或整合的算法。从论文题目也可以看出,PASTE提供了两种模式,即数据对齐(alignment)和数据整合(integration)。
后面我将分别介绍这两种方法。
2.1 数据预处理
在介绍两种模式之前,我要先介绍数据预处理方式并以及明确一下符号定义,方便大家理解后面对于两种模式具体实现方法的介绍。
ST实验得到的结果数据是一对矩阵(X, Z),其中是一个p*n的矩阵,p为基因数量,n为捕获区的点数,
代表第j个点处第i个基因的表达水平(数量),
向量为第j个点的基因表达水平。矩阵Z是一个2*n矩阵,其中第j列的二维向量
代表第j个点在捕获区的二维坐标,即空间位置。
由于在进行ST实验时组织切片放置方向比较随机,因此直接使用Z矩阵作为空间位置并不高效,因此使用相对位置矩阵D,是一个n*n的矩阵,
为第i个点与第j个点的相对位置。
此外,对每一个点i,设置一个严格大于零的权重用于表示该点相对于其他点的重要程度,并且有限制
。如果没有先验知识指导权重的设置,那么就令每个点的权重相同。
设置代价函数,该函数用于衡量两个点处基因表达水平的相似程度,例如使用KL散度,应该越相似函数值小。
那么现在的两个切片数据就变为(X, D, g)和(X', D', g'),其中两个切片分别具有n和n'个点,点数不一定相同是因为只考虑捕获区接触到组织切片的点。
2.2 数据对齐(pairwise slice alignment)
2.2.1 对齐目的
第一种数据处理模式是“对齐”(pairwise slice alignment)。
假设我们现在有两个切片中获得的数据,每个切片可以按照上图中左侧中间的点阵理解,其中正方形点阵为整个捕获区,蓝色点为捕获区中未接触到组织切片的部分,绿色点为捕获区中接触到组织切片的部分。
对于每一个点,可以捕获到几个到几十个细胞。因此对于每一个点,我们知道的基因表达水平信息是这个点接触到的所有细胞的一个综合信息,而空间信息体现在每个点在这个点阵中的位置。
从左上角的图像我们可以知道,这些组织切片之间具有垂直的空间关系,而我们现在具有的只是每个切片分别获取的数据,因此“对齐”这个模式的目的是将现有的两个切片上的点进行对齐,使其能够恢复出原来的垂直位置关系。
注意这里我们并不是想要将两个切片中的单个点与单个点直接进行对齐,即切片A中的点i,不一定只能对应切片B中的点j,尽管这可能更符合一般的理解。在实际计算中,由于数据缺失等各种原因,很可能切片A中的点数与切片B中的电点数不相同,盲目将两个切片的点进行1对1的对应并不可取。实际上可以将两个切片之前看成一个全连接层,切片A中的任一点都与切片B中的每个点存在一个权重,当然,最后计算出来找到最优的权重后,也会出现切片A中的点i,与切片B中某一点j的权重明显高于其他点的一种稀疏的结果。
每次只对两个切片进行对齐,如果有一系列切片A、B、C……,那么就可以按照A&B、B&C……的方式依次对齐,最终可以通过这些切片恢复出组织的3D结构。
2.2.2 具体实现方法
下面我们来看一下“对齐”模式的具体实现。
“对齐”就是要找到一个两个切片之间的概率映射,
为n*n'的矩阵,满足条件
,并使其具有下述两种性质:
1)如果切片A中的点i与切片B中的点j间具有较大权重,那么点i的基因表达水平
与点j的基因表达水平
相近。
2)如果切片A中的点对(i, k)与B中的点对(j, l)分别具有较高权重和
,那么i与k在A中的空间距离
应该与j和l在B中的空间距离
相近。
这两点分别考虑了基因表达水平的相似程度和空间位置的相似程度,这也正是PASTE算法的精髓所在。
我们通过最小化下面的式子来找到概率映射:

该式子为两部分相加的形式,第一部分表示基因表达水平的相似程度信息,相似程度越高,代价c函数值越小,第二部分用于表示空间位置的相似程度,相似程度越高,两个距离之差的平方越小。其中α是一个超参数,用于控制对于基因表达水平和空间位置的权重分配。
2.3 数据整合(center slice integration)
2.3.1 整合目的
第二种数据处理模式是“整合”(center slice integration)。
“整合”是将多个ST切片整合为单一的“中心切片”,该中心切片与独立切片在基因表达水平和空间关系上具有高度的相似性,并且该由于细胞类型和状态有限且较少,因此中心切片具有低秩性。
其目的是克服由于不同的测序覆盖、组织解剖或捕获区上的组织放置而导致的单个切片的可变性。
论文中提到,由于多数现有的ST数据集中,每个组织切片的厚度为10~20μm,这小于捕获区点的直径(例如在10x Visium中是55μm)和点与点之间的间距(例如在10x Visium中是100μm),因此对于这样的数据集,通过多切片整合对于下游分析带来的优势要超过放弃获得3D结构造成的劣势。
2.3.2 具体实现方法
下面我们来看一下“整合”模式的具体实现。
数据整合的实现基于以下三点假设:
1)给出的不同ST切片之间非常相似,因此可以整合成一个单一中心切片(由于不同组织切片之间的垂直间距很小,因此这一点可以被满足)。
2)空间坐标和中心切片上的点的分布是事先已知的(由于捕获区域中点的位置由技术确定,因此这一点可以被满足)。
3)基因表达水平矩阵是低秩的(由于细胞类型和状态有限且较少,因此这一点可以被满足)。
现有t个切片数据,每个切片上的点数为
,我们的目标是计算出中心切片
。现在需要额外添加参数
,并认为X具有非负矩阵分解X=WH,其中W为p*m为矩阵,H为m*n维矩阵,m为正整数。

通过最小化上述函数,求得W和H,以及,其中
为中心切片到第i个切片的概率映射。
具体求解方式使用坐标下降法,轮流执行步骤1(令W和H不变,优化的)和步骤2(令
不变,优化W和H)。
3 改进方向
PASTE算法仍有一些不足,作者在结尾给出了一些改进方向:
1)PASTE不使用通常伴随ST数据的组织学图像,通过利用组织学图像和使用图像配准领域的方法可以进一步改进PASTE。
2)时间开销较大,可以使用GPU加速或使用小批量或子集数据。
3)调整PASTE优化的目标函数,考虑在空间结构和细胞组成方面存在显著差异的切片。从而使PASTE可以应用于来自不同患者的空间转录组实验数据,以找到在不同患者之间存在的基因表达空间模式关联。
4)文中将PASTE应用于10x Genomics技术获取的数据中,而PASTE还能够应用于其他空间技术(如smFISH,seqFISH+,STARmap和Slide-Seq2)获取的数据。
5)开发改进的ST数据模拟器将有助于模拟真实数据,如组织的挤压或拉伸。















 1449
1449











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









