







 本文详细介绍了使用Plink进行遗传数据分析的质量控制步骤,包括个体和SNP缺失率筛选、性别质控、MAF过滤、哈温平衡检验、杂合率检验以及去除亲缘关系近的个体。此外,还探讨了群体结构分层和关联分析方法,如阈值性状的assoc和logistic分析,以及假阳性控制策略。
本文详细介绍了使用Plink进行遗传数据分析的质量控制步骤,包括个体和SNP缺失率筛选、性别质控、MAF过滤、哈温平衡检验、杂合率检验以及去除亲缘关系近的个体。此外,还探讨了群体结构分层和关联分析方法,如阈值性状的assoc和logistic分析,以及假阳性控制策略。
目录
0 准备
参考
https://zhuanlan.zhihu.com/p/126178268
GWAS教程
https://onlinelibrary.wiley.com/doi/full/10.1002/mpr.1608
下载项目和数据
ssh software-install
git clone https://github.com/MareesAT/GWA_tutorial/
cd GWA_tutorial
ll
plink可以读取文本格式的文件或二进制文件,由于读取大型文本文件可能很耗时,因此建议使用二进制文件。
文本plink数据包含两个文件:
- 包含有关个体及其基因型的信息(*.ped);
- 包含有关遗传标记的信息(*.map)
二进制plink数据由三个文件组成:
- 包含单个标识符(ID)和基因型(*.bed)
- 包含有关个体(*.fam)
- 遗传标记(*.bim)

二进制格式转文本格式,生成test.map test.ped:
plink --bfile HapMap_3_r3_1 --recode --out test1 遗传数据的质量控制
数据来源 http://hapmap.ncbi.nlm.nih
数据下载 https://github.com/MareesAT/GWA_tutorial/ (1_QC_GWAS.zip)
unzip 1_QC_GWAS.zip
cd 1_QC_GWAS
ll
质量控制步骤
| 步骤 |
命令 |
功能 |
阈值和解释 |
| 1: SNP和个体的缺失 |
--geno |
排除了大部分受试者中缺失的SNP。在此步骤中,低基因型的SNP被删除。 |
建议首先以宽松的阈值(0.2; > 20%)过滤SNP和个体,从而过滤掉缺失程度很高的SNP和个体。再使用更严格的阈值过滤(0.02)。 |
|
|
--mind |
排除基因型缺失率高的个体。在此步骤中,低基因型的个体被移除。 |
注意:在个体移除前应先进行SNP过滤。 |
| 2. 性别差异 |
--check-sex |
根据X染色体杂合/纯合率检查数据集中记录的个体性别与他们自身性别的差异。 |
可以指示样品混合。如果许多研究对象都存在这种差异,则应仔细检查数据。男性的X染色体纯合度估计值为0.8,女性为0.2。 |
| 3. 最小等位基因频率(MAF) |
--maf |
仅包括超过MAF阈值的SNP。 |
低MAF的SNP很少见,因此缺乏检测SNP表型关联的能力。这些SNP也更容易出现基因分型错误。MAF阈值应取决于您的样本大小,较大的样本可以使用较低的MAF阈值。对于大样本(N = 100.000)与中等样本(N=10000),通常采用0.01和0.05作为MAF阈值。 |
| 4. 哈迪-温伯格平衡(HWE) |
--hwe |
排除偏离哈代-温伯格平衡的指标。 |
基因分型错误的常见指标也可能表明进化选择。 对于二元性状,建议排除:病例中HWE p值<1e-10以及对照组中<1e-6。不大严格的病例阈值可避免丢弃在选择下与疾病相关的SNP。 对于数量性状,推荐HWE p值<1e-6。 |
| 5. 杂合性 |
|
排除杂合率高或低的个体 |
偏差可能表明样品受到污染,近亲繁殖。 建议删除样本杂合率均值偏离±3标准差的个体。 |
| 6. 相关性 |
--genome |
通过下降(IBD)计算所有样本对的同一性。 |
使用独立的SNP(剪枝)进行分析,并将其限制为仅常染色体。 |
|
|
--min |
设置阈值,并创建一个关联性高于所选阈值以上的个体列表。这意味着可以检测到如pihat>0.2的有亲属关系的受试者(即二度亲属)。 |
隐性关联会干扰关联分析。如果你有一个以家庭为基础的样本(例如:父母-后代),则不需要删除相关对,但统计分析应考虑到家庭的相关性。然而,对于基于人群的样本,建议使用pihat阈值为0.2。 |
| 7. 人口分层 |
--genome |
通过下降(IBD)计算所有样品对的同一性。 |
使用独立的SNP(剪枝)进行分析,并将其限制为仅常染色体。 |
|
|
--cluster –mds-plot k |
基于IBS生成数据中任何子结构的k维表示。 |
k是需要定义的维数(通常为10)。这是QC的一个重要步骤,它包含多个过程,但为完整起见,此步骤将在“控制人口分层”一节中更详细地描述。 |
plink数据格式转化
plink --bfile HapMap_3_r3_1 --recode --out test输出文件:test.map test.ped
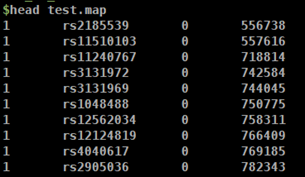

查看基因型个体和SNP数量
wc -l test.map test.ped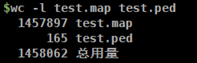
可以看出共有165个基因型个体,共有1457897个SNP数据。
查看个体缺失的位点数,每个SNP缺失的个体数
plink --bfile HapMap_3_r3_1 --missing
输出plink.imiss和 plink.lmiss两个文件。
- plink.imiss:个体缺失位点的统计
- plink.lmiss:单个SNP缺失的个体数
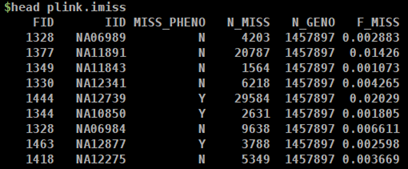
【个体缺失位点统计】
第一列为家系ID,第二列为个体ID,第三列是否表型缺失,第四列是缺失的SNP个数,第五列总SNP个数,第六列缺失率。
【SNP缺失的个体数】
第一列为染色体,第二列为SNP名称,第三列为缺失个数,第四列为总个数,第五列为缺失率。

缺失结果可视化
Rscript --no-save hist_miss.R# hist_miss.R
indmiss<-read.table(file="plink.imiss", header=TRUE)
snpmiss<-read.table(file="plink.lmiss", header=TRUE)
# read data into R
pdf("histimiss.pdf") #indicates pdf format and gives title to file
hist(indmiss[,6],main="Histogram individual missingness") #selects column 6, names header of file
pdf("histlmiss.pdf")
hist(snpmiss[,5],main="Histogram SNP missingness")
dev.off() # shuts down the current device【单个位点缺失率统计】

【基因型个体缺失率统计】

1.1 对个体及SNP缺失率进行筛选
以宽松的阈值(0.2; > 20%)过滤SNP和个体,从而过滤掉缺失程度很高的SNP和个体。再使用更严格的阈值过滤(0.02)。
- 如果一个SNP在个体中2%都是缺失的,则删除该SNP,参数为 --mind 0.02
- 如果一个个体,有2%的SNP都是缺失的,则删除该个体,参数为 --geno 0.02
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









