







转录调控涉及调控序列(分子序列,比如启动子,增强子等)和蛋白质之间的复杂相互作用,指导所有生物过程。建模转录的计算模型缺乏通用性,无法准确推断到未知的细胞类型和条件。GET(通用表达Transformer)是一个可解释的基础模型,揭示了 213 种人类胎儿和成人细胞类型中的调控语法。GET 完全依赖染色质可及性数据和序列信息,即使在以前未见过的细胞类型中也能达到实验级的基因表达预测准确度。GET 还在新的测序平台和检测中表现出显著的适应性,能够在广泛的细胞类型和条件下进行调控推断,并揭示通用和细胞类型特异性的转录因子相互作用网络。总体而言,GET提供了一个通用且准确的转录模型,以及具有细胞类型特异性的基因调控和转录因子相互作用目录。
原文请参考:Fu, X., Mo, S., Buendia, A. et al. A foundation model of transcription across human cell types. Nature (2025). https://doi.org/10.1038/s41586-024-08391-z
背景概述
转录调控支撑着各种生物过程。转录变化由保守的调控机制协调,包括与调控序列结合的转录因子 (TF)、辅激活因子、介质和核心转录因子以及 RNA 聚合酶 II (PolII)。已知 TF 结合位点基序的聚集表明 TF DNA-binding domain具有显著的同源性,这降低了调控相互作用的组合变异。然而,人们对转录调控的理解通常仅限于特定的细胞类型,并且不清楚不同 TF 的组合相互作用如何决定在细胞类型中观察到的表达谱的多样性。以前的表达预测方法如 Expecto、Basenji 和 Enformer 旨在对训练细胞类型进行微调后进行预测,从而阻碍了这些模型的通用性。
最近,GPT-4 和 ESM-2 等基础模型已成为一种变革性方法。通过对广泛而多样的数据集进行预训练,基础模型提供了对大规模语料库的广义理解,在此基础上可以构建专门的适应性以解决特定任务或挑战。例如,Geneformer、scGPT 和 scFoundation 等单细胞基础模型的最新发展已经证明了如何在一个模型中编码不同的转录组谱,从而实现各种下游任务,包括细胞类型注释和扰动预测。然而,目前还没有探索转录如何从染色质景观中出现的基础模型。
作者介绍了通用表达Transformer (GET),这是一种可解释的转录调控基础模型。GET 从 213 种人类胎儿和成人细胞类型的染色质可及性数据中学习转录调控语法,并准确预测seen和unseen细胞类型中的基因表达 (图 1a)。此外,GET 提供报告基因检测读数的零样本预测,在识别顺式调控元件方面优于以前最先进的模型,并识别以前未知和已知的胎儿血红蛋白上游调节因子。GET 还提供了丰富的细胞类型特异性调控见解:利用 GET 预测的共调节信息,可以精确定位潜在的基序-基序相互作用并构建了Human TF 和辅激活因子的结构相互作用目录 (图 1b)。利用这一点,GET确定了涉及 PAX5和核受体家族 TF 的淋巴细胞特异性 TF-TF 相互作用,并强调了影响 PAX5 无序区域与核受体结构域结合的白血病相关种系变异的疾病机制。
PAX5是一种TF,属于PAX家族中的一员,调控B细胞相关的基因表达,PAX5通过结合特定的DNA序列(即PAX5结合位点)来调控基因表达。
PAX5 无序区域:Intrinsically Disordered Region, IDR通常是没有固定三维结构的蛋白质区域,在不同环境中具有不同构象。
核受体结构域:位于细胞核的蛋白质,能够直接结合到DNA并调控基因表达。
GET输入:peak (可访问区域) × TF (基序) 矩阵,表示在特定peak位置上检测到TF基序的强度或者存在情况。
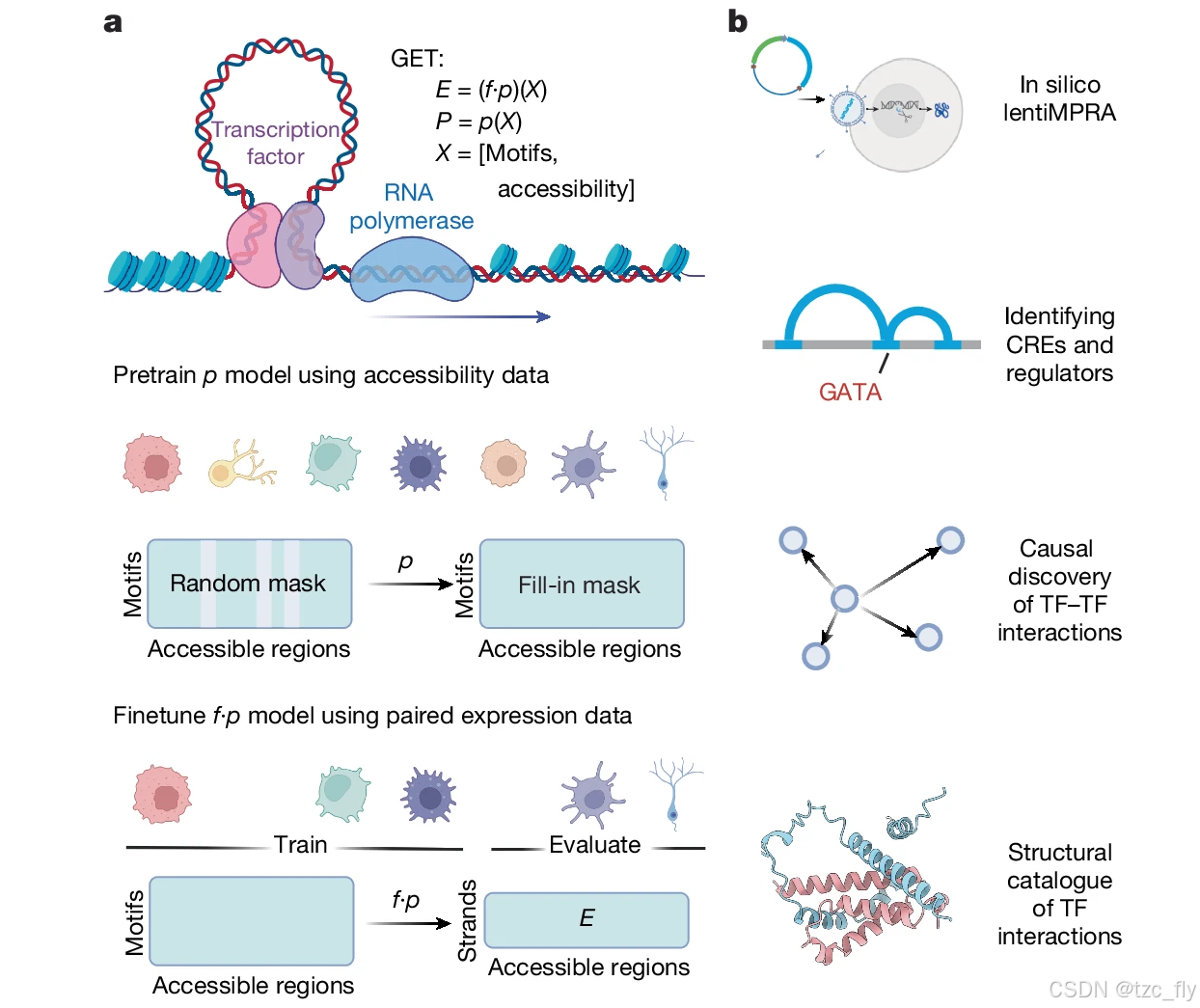
- 图1a:GET 示意图。GET 的输入是来自Human单细胞 scATAC-seq 图谱的peak (可访问区域) × TF (基序) 矩阵。通过对超过 200 种细胞类型的输入数据进行自监督随机mask预训练,GET 学习了转录调控语法 ( p p p)。GET 在配对的 scATAC-seq 和 RNA-seq 数据上进行微调,并学习将调控语法转化为基因表达,即使在unseen的细胞类型中也是如此 ( f ⋅ p f⋅p f⋅p)。
- 图1b:GET 下游应用示意图。CRE表示顺式调控元件(cis-regulatory element)。
ATAC-seq数据预处理
Pseudobulk
为了确定每个区域的染色质可及性得分,作者使用了来自各个研究的 scATAC-seq 计数表和细胞注释表。为了将单个细胞聚合成“伪bulk细胞类型”,作者使用了来自每个研究的 Louvain 聚类结果。每个伪bulk使用Log count per million (logCPM)。每个簇的注释用于确定生物细胞类型。根据经验,设置了超过 600 个细胞计数的阈值,以确保选定细胞簇的测序深度足够。数据仅以伪bulk格式呈现。所有细胞类型都是来自正常组织的细胞类型。预训练数据集中未包含任何疾病状态。直到在下游任务中才加入了进一步的数据集,例如 K562 和肿瘤细胞的零样本分析。
细胞类型特异的可及性区域识别
为了编制每个细胞类型特有的可访问区域列表,作者过滤掉了没有计数的峰值。在人类胎儿和成人染色质可访问性图谱的背景下,使用了 Zhang 等人制作的峰值集,结合了 Domcke 等人最初发表的胎儿染色质可访问性图谱,还使用 Domcke 等人的原始峰值调用和细胞类型注释训练了仅限胎儿的 GET 模型版本。对于 10×multiome 数据,作者使用了提供的峰值片段计数矩阵。对于 K562 NEAT-seq 和bulk染色质可访问性数据,使用 MACS2调用了更宽松的峰值版本,并对生成的峰值集应用了不同的 logCPM 截止值以选择可访问区域。
可及性特征
在该研究中,特定基因组区域的染色质可及性得分由给定细胞类型伪bulk中位于该区域内的片段数定义。为了增强模型的通用性,这些计数通过 logCPM 程序进一步标准化。具体而言,设 t t t 为伪bulk中的总片段数,设 c i c_{i} ci 为区域 i i i 中的片段数。然后,可及性得分 s i s_{i} si 可以计算如下: s i = l o g 10 ( c i t + 1 ) , t = ∑ i c i s_{i}=log_{10}(\frac{c_{i}}{t}+1), \thinspace\thinspace t=\sum_{i}c_{i} si=log10(tci+1),t=i∑ci对于大多数调控分析,GET 模型的二元 ATAC 版本用于全面评估 TF 施加的调控影响。在此特定模型版本的训练和推理阶段,如果该区域被识别为染色质可及性峰,则可及性分数统一设置为 1。这相当于在研究场景中假设二元染色质可及性状态。
Motif特征
为了计算特定基因组区域内的基序(motif)结合得分,使用 Vierstra 等人先前编译的 2,179 个 TF 基序位置权重矩阵(可在 https://www.vierstra.org/resources/motif_clustering 上访问)扫描 hg38 参考基因组中的相应序列。对于扫描过程,使用 MOODS 工具和默认阈值。
预训练中使用的 213 种细胞类型的注释遵循胎儿和成人可及性图谱中提供的原始细胞类型分类。该分类是通过对 ATAC-seq 计数谱进行聚类实现的,随后根据已知marker基因的表达进行标记。组织和细胞类型的综合列表及其注释可在人类细胞图谱 (http://catlas.org/humanenhancer/#!/cellType) 中找到。原始图谱包括 222 种胎儿和成人细胞类型,但经过进一步过滤,删除了测序覆盖率低的细胞类型。这种方法确保模型在不同的细胞环境中进行训练,同时确保染色质可及性伪bulk的覆盖率足够。
输入数据
GET 旨在捕捉不同区域和调控子regulators之间的相互作用。为了实现这一点,我们需要每个输入样本包含一定数量的连续可访问区域。通过之前的实验,作者发现理想的等效基因组覆盖范围约为 2 Mbp 或更大,大多数染色质接触都发生在这个范围内。因此,基于目前的数据预处理流程,作者选择每个训练样本使用 200 个输入区域。在预训练期间使用非重叠采样,在微调期间使用滑动窗口方法。滑动窗口的步幅设置为一个样本中区域数量的一半(即,对于具有 200 个峰的样本,每步 100 个峰)。
选择每个样本的区域数量是为了在计算效率和包含每种细胞类型的调控景观代表性样本的需求之间取得平衡。值得注意的是,这 200 个peak所覆盖的实际基因组跨度可能因多种因素而异,包括染色质可及性的细胞类型特异性变化、峰调用期间应用的阈值以及峰的染色体分布。在涵盖胎儿和成人细胞类型的统一处理的数据集中,我们观察到 200 个峰通常对应于大约 2-4 Mbp 的基因组范围。这一估计源于这样的理解:人类基因组有大约 30 亿个碱基对,全面分析后可产生大约 150,000 个可访问的峰。因此,考虑到峰在不同细胞类型中的分布和密度,200 个峰平均代表 2-4 Mbp 的基因组跨度。在训练模型时,边界完全取决于采样峰的边界。
每个输入样本通常包含大约200个连续的调控区域(peaks),这些区域覆盖约2-4兆碱基对(Mbp)的基因组范围,对于每个调控区域,扫描DNA序列以识别给定转录因子结合位点。所以,每个样本格式都是(peaks,motifs),peaks 200个,motifs数量由给定的TF数量确定。
RNA-seq数据预处理
对于涉及多组学的实验,可及性和表达之间的对应关系本质上是通过细胞barcode确定的。作者将表达数据映射到调控区域中,做法是把基因表达值分配到输入样本的200个调控区域中。与 200 × 283 输入矩阵一致,target输入是 200 × 2 矩阵,表示正链和负链中相应 200 个区域的转录水平。
对于如何确定基因表达该分配到正链还是负链:
- 将RNA-seq读取比对到参考基因组时,比对软件(如STAR、Hisat2)会记录每个读取的比对方向。根据比对结果,可以确定哪些读取是从正链转录的,哪些是从负链转录的。
模型架构
GET 架构由三部分组成:(1) 调控元件嵌入层 (RegionEmb);(2) 调控元件注意层 (Encoder);(3) 线性输出层作为表达预测头,或者其他输出头。GET 将 200 个调控元件(每个元件有 282 个基序结合分数和一个可选的可及性分数)作为输入样本。因此,输入是一个 200 × 283 矩阵。当选择不使用定量可及性分数时,将第 283 列中的值设置为 1。
将样本输入到 RegionEmb 层,为每个peak生成维度为 768 的调控元件嵌入。由于不想丢失原始调控元件输入中的信息,应用线性层来捕获不同类别的 TF 结合位点中的一般信息。为了了解调控元件和 TF 之间的顺式和反式相互作用,作者在调控元件嵌入上应用了 12 个编码器层和多头注意 (MHA) 机制。
假设
N
h
N_h
Nh、
d
v
d_v
dv 和
d
k
d_k
dk 分别表示 head 的数量、value 的深度和 key 的深度。每个 head
h
h
h 的输出计算如下:

其中,
W
q
,
W
k
∈
R
(
n
×
D
)
×
d
k
W_{q},W_{k}\in\mathbb{R}^{(n\times D)\times d_{k}}
Wq,Wk∈R(n×D)×dk,
W
v
∈
R
(
n
×
D
)
×
d
v
W_{v}\in\mathbb{R}^{(n\times D)\times d_{v}}
Wv∈R(n×D)×dv是线性参数。然后,将每个头
h
h
h 的输出连接到调控元件注意块。最后使用层规范化 (LN)、前馈网络 (FFN) 和残差连接来生成每个层的输出:

其中,
z
l
′
z_{l}'
zl′,
z
l
−
1
z_{l-1}
zl−1为block
l
l
l的中间表示,
z
l
−
1
z_{l-1}
zl−1为block
l
−
1
l-1
l−1的输出。
GET 架构与最先进的模型 Enformer 类似。但是,以下改进超越了该模型的性能:GET 使用调控元件嵌入层来捕获不同类别的 TF 结合位点中调控元件的一般信息。此外,使用mask调控元件机制来学习来自不同人类细胞类型的调控元件与 TF 之间的一般顺式和反式相互作用。具体而言,统一选择一组随机位置进行掩蔽,掩蔽率为 0.5。
作者对大规模单细胞染色质可及性数据进行了预训练。然后,使用与 Enformer 相同的泊松负对数似然损失函数,在配对染色质可及性-基因表达数据上对预训练模型进行微调。
建模细胞类型特异表达
GET 的设计理念植根于转录调控机制。具有启动子和调控元件的局部基因组区域(约 2 Mbp)可以通过这些元件与不同 TF 的结合程度以及它们在特定细胞类型中的可访问性来表征。这些特征塑造了染色质环境 (p(X)),该环境控制 PolII 如何驱动每个单独元件的表达,这可以从 RNA 测序 (RNA-seq) 数据中近似得出。使用专门为调控元件设计的嵌入和注意架构,进行了自监督预训练,以允许 GET 了解区域和特征如何在不同细胞类型中相互作用。具体而言,通过随机屏蔽调控元件,模型被训练以预测被屏蔽区域中的基序结合分数和可选的可访问性分数。随后,PolII 将把染色质环境 p(X) 读出到表达式 E 中。具有相同架构但不同输出头的微调阶段模拟了此过程(图 1a 和扩展数据图 1)。
这种两阶段设计使得无需配对表达测量即可使用染色质可及性数据,从而大大提高了训练数据中调控信息的多样性。GET 的预训练使用从 213 种人类胎儿和成人细胞类型的scATAC-seq数据的单细胞测定中收集的伪bulk染色质可及性。其中,153 种细胞类型与通过多组学方案或单独的单细胞 RNA-seq 实验获得的表达数据相结合,并用于微调和评估。

- 扩展图1:GET 模型使用染色质可及性数据来预测基因表达。输入数据由 ATAC-seq 峰的 DNA 序列组成,表示为 (B, R, L*, 4),其中 B 是batch大小,R 是区域数,L* 是可变序列长度,4 代表四个 DNA 碱基。基序映射将序列转换为基序矩阵 (B, R, M),其中 M 是基序数。定量 ATAC 信号可以选择性地包含在 M 中。RegionEmb 层将基序矩阵投影到潜在空间 (B, R, D),其中 D 是潜在维度。然后由 12 个 Transformer 编码器层处理此嵌入。模型输出由投影层生成,然后进行 SoftPlus 激活,产生 (B, R, O) 张量,其中 O 是输出通道。
- 图中显示了四个输出头:MaskHead:在预训练期间预测掩蔽基序矩阵元素 (O = (R/2, M))。 ExpHead:预测滞留基因表达为 log1p(TPM) (O = 2)。ATACHead:预测峰值可及性为 log1p(CPM) (O = 1)。CAGEHead:预测峰值 CAGE 信号为 log1p(CPM) (O = 1)。该模型架构能够灵活处理调控基因组数据,捕捉调控元件和转录因子之间的复杂相互作用,以预测各种基因组特征和基因表达水平。
作者首先评估了 GET 在表达微调过程中遗漏一种细胞类型的环境中准确预测未见细胞类型中基因表达的能力。对于遗漏的星形胶质细胞和具有可接近启动子的基因,GET 预测表达值与观察到的表达之间的 Pearson 相关性达到 0.94(R2 = 0.88)(图 1c)。GET 的性能超过了转录起始位点 (TSS) 可接近性(r = 0.47,R2 = -0.23)和基因活性评分(r = 0.51,R2 = -0.67),这强调了 DNA 序列特异性和远端上下文信息在转录调控中的重要性。此外,GET 的表现优于最高相关细胞类型表达 (r = 0.83,R2 = 0.62) 和训练细胞类型中的平均表达 (r = 0.78,R2 = 0.53;图 1c)。

- 图1c:对未见细胞类型(胎儿星形胶质细胞)的 GET 预测性能基准。每个点代表一个基因。颜色表示 TSS 中标准化的染色质可及性。基因活性是现代 scATAC-seq 分析流程中广泛使用的分数。相关性最高的细胞类型是其观察到的基因表达与胎儿星形胶质细胞(在本例中为胎儿抑制神经元)相关性最强的训练细胞类型。平均细胞类型是跨训练细胞类型观察到的平均基因表达。虚线表示线性拟合。对星形胶质细胞中所有可及的 TSS 进行预测,并平均到基因水平。
识别顺式调控元件
通过模型解释技术,可以有效地得出跨细胞类型具有可访问启动子的基因的区域或基序贡献分数。作者专注于胎儿红细胞,利用已发表的基因组碱基编辑数据来研究四个已知的胎儿血红蛋白调节基因座(BCL11A、NFIX、KLF1 和 HBG2;已知前三个调节胎儿血红蛋白,HBG2 编码胎儿血红蛋白亚基;图 3a)。

- 图3a:案例研究确定了控制表型、胎儿血红蛋白 (HbF) 水平的顺式调节元件和调节器。
将 GET 应用于胎儿红细胞,可以深入了解胎儿血红蛋白的调节。作者重新发现了 GATA TF 的核心作用,它通过与红细胞特异性增强子结合,协调 BCL11A 的表达,BCL11A 是一种已知的血红蛋白调节调节剂。GET 还强调了 SOX 家族 TF 在该增强子中的作用,该家族之前与胎儿血红蛋白有关,但尚不清楚其是否通过该特定增强子发挥作用(图 3b)。

- 图3b:GET 识别出红细胞特异性增强子中的 GATA 基序,该增强子上调 BCL11A(一种 HbF 阻遏物)。顶部,向导 RNA (gRNA) 富集分数 (HbFBase);分数越高表示在更多 HbF 细胞中富集,这意味着这些编辑会干扰可以上调 BCL11A 的顺式调节元件或调节器结合位点。中间,来自胎儿红细胞的 scATAC-seq 信号和峰值。底部,红细胞特异性增强子中 BCL11A 表达的基序贡献分数。
检查所有四个基因座 - BCL11A、NFIX、KLF1 和 HBG2。作者将 GET 与已建立的模型(包括 Enformer、HyenaDNA、DeepSEA 和 Activity-by-Contact (ABC))进行了基准测试。 GET 的表现优于这些同类方法,尤其是在检测长距离增强子-启动子相互作用方面(图 3c-d )。

- 图3c:基因组轨迹显示推断的 BCL11A 和 NFIX 基因座顺式调控元件。从上到下,轨迹代表:HbFBase,显示来自碱基编辑实验的 gRNA 富集分数;GET,显示推断的区域重要性分数;Enformer,显示推断的区域重要性分数;HyenaDNA,显示使用预训练的 HyenaDNA 语言模型的计算机诱变 (ISM) 结果;ABC Powerlaw,显示使用胎儿红细胞 ATAC 和 K562 Hi-C 幂律的接触活性预测;来自红细胞系 HUDEP-2 的 ATAC-seq 数据;来自胎儿红细胞的 ATAC-seq 数据,用于训练 GET;以及来自 HUDEP-2 的 HiChIP-seq 数据,展示染色质相互作用。
- 图3d:将 GET 与其他增强子-启动子对预测方法进行比较的基准测试结果,包括对远端(大于 100 kb)相互作用的分析。顶部,红细胞胎儿血红蛋白调节增强子预测。底部,K562 CRISPRi 增强子靶标预测;显示精确度-召回率曲线下面积 (AUPRC)。图中还显示了不同 GET 预测成分(Jacobian、DNase、Powerlaw;方法:“增强子-基因对预测”)的消融。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









