










1. 背景知识
1.1 生物背景
人类基因组、基因、外显子、内含子、CDS、UTR、中心法则
1.2 癌症背景
突变:SNP、SNV、CNV、SV、somatic mutations、germline mutations
癌症相关基因:原癌基因、抑癌基因
突变类型:驱动突变、乘客突变、克隆与亚克隆、主干突变、分支突变、私有突变
肿瘤进化、肿瘤微环境、TMB、肿瘤异质性、肿瘤基因组不稳定性
1.3 测序背景
NGS测序原理,测序读长、深度、质量,全基因组测序WGS,全外显子测序WES,靶向测序panel
1.4 数据库背景
UCSC、NCBI、Ensemble、GATK、COSMIC、1000g、dbsnp、SRA
1.5 文件格式
fasta,fastq,sam,bam,bed,gtf,gff,vcf
2. 搭建分析环境
创建文件夹存放各种软件包、数据库文件以及分析结果
## 首先在用户的主目录下创建 wes_cancer 文件夹作为工作目录
mkdir ~/wes_cancer
cd ~/wes_cancer
## 在 ~/wes_cancer 中创建 biosoft project data 三个文件夹
## biosoft 存放软件安装包
## project 存放分析过程产生的文件
## data 存放数据库文件
mkdir biosoft project data
cd project
## 在 project 文件夹中创建若干个文件夹,分别存放每一步产生的文件
mkdir -p 0.sra 1.raw_fq 2.clean_fq 3.qc/{raw_qc,clean_qc} 4.align/{qualimap,flagstat,stats} 5.gatk/gvcf 6.mutect 7.annotation/{vep,annovar,funcotator,snpeff} 8.cnv/{gatk,cnvkit,gistic,facet} 9.pyclone 10.signature
2.1 安装软件
使用conda安装大部分软件可以解决大部分的软件依赖问题,对新手相较友好。
2.1.1 安装conda
进入清华镜像源:https://mirror.tuna.tsinghua.edu.cn/help/anaconda/,复制Miniconda的链接地址
## 下载
cd ~/wes_cancer/biosoft
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
## 安装
bash Miniconda3-latest-Linux-x86_64.sh
## 安装过程中如果提示要输入 yes/no,就选 yes;如果有提示要按 Enter,那就按 Enter 回车键
## 重新激活环境
source ~/.bashrc
## 关闭conda自启动
conda config --set auto_activate_base false
## 添加清华镜像
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
conda config --set show_channel_urls yes
2.1.2 用conda安装软件
新建分析环境wes,并激活,然后安装生信软件以及必要的团建
## 新建小环境 wes
conda create -n wes python=3
## 激活 wes 小环境
conda activate wes
## 安装必要的生信软件
conda install -y sra-tools fastqc trim-galore multiqc bwa samtools gnuplot qualimap subread vcftools bedtools cnvkit
conda install -y -c hcc aspera-cli=3.7.7
2.1.3 安装GATK
进入GATK官网https://gatk.broadinstitute.org/,找到最新安装包,复制链接地址。
cd ~/wes_cancer/biosoft
wget -c https://github.com/broadinstitute/gatk/releases/download/4.1.9.0/gatk-4.1.9.0.zip
unzip gatk-4.1.9.0.zip
2.2 下载数据库文件
2.2.1 人类参考基因组hg38
测序之后拿到的数据是 fastq 文件,是记录了 reads 及测序的质量值,我们需要比对到参考基因组上才能让这些数据有意义,因此我们需要下载参考基因组文件,用到的是 hg38 版本。因为我们后面要用到 gatk ,而 gatk 对参考基因组有一定的要求,需要下载 gatk 指定的参考基因组。
GATK --> Resource Bundle --> https://gatk.broadinstitute.org/hc/en-us/articles/360035890811-Resource-bundle --> 数据
ftp服务器网址:ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/
cd ~/wes_cancer/data/
wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Homo_sapiens_assembly38.fasta.gz
2.2.2 gatk需要用到的其他文件
其实 gatk 要下载的文件有很多,所以我们可以使用 nohup…& 的形式将下载的命令都提交到后台(如果网络不好,可能会下载失败,所以下载后请自行检查)。
## gatk
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.dbsnp138.vcf &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.dbsnp138.vcf.idx &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta.fai &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.dict &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/1000G_phase1.snps.high_confidence.hg38.vcf.gz &
nohup wget -c https://console.cloud.google.com/storage/browser/_details/genomics-public-data/resources/broad/hg38/v0/1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/funcotator/funcotator_dataSources.v1.6.20190124s.tar.gz &
## gatk_ftp
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/dbsnp_146.hg38.vcf.gz &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/dbsnp_146.hg38.vcf.gz.tbi &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Homo_sapiens_assembly38.fasta.gz &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Homo_sapiens_assembly38.fasta.fai &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Homo_sapiens_assembly38.dict &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi &
nohup wget -c ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/funcotator/funcotator_dataSources.v1.6.20190124s.tar.gz &
2.2.3 其他数据库文件
除GATK流程分析所需的数据,还需要下载参考基因组的注释文件,通常是gtf格式,可以到ensemble或genecode数据库下载
## bed
wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/CCDS.current.txt
cat CCDS.current.txt | grep "Public" | perl -alne '{/\[(.*?)\]/;next unless $1;$gene=$F[2];$exons=$1;$exons=~s/\s//g;$exons=~s/-/\t/g;print "$F[0]\t$_\t$gene" foreach split/,/,$exons;}'|sort -u |bedtools sort -i |awk '{if($3>$2) print "chr"$0"\t0\t+"}' > hg38.exon.bed
3. 读文献并下载数据
3.1 下载SRA数据
3.1.1 官网下载
NCBI 的 Sequence Read Archive(SRA),每个项目的 url 格式都是一样的。

进入官网根据SRA编号下载:https://www.ncbi.nlm.nih.gov/sra/?term=,点击 Metadata 和 AcessionList 下载两个文件,存放至 ~/wes_cancer/project 目录下。

然后从 SRA_Acc_List.txt 文件中拿到 id 号并用 sra-tools 的 prefetch 命令下载 sra 文件。
cat SRR_Acc_List.txt | while read id
do
prefetch ${id} -O ./0.sra
done
下载完成后,在 0.sra 目录下,每个 sra 文件以单独的文件夹的形式存放,每一个 sra 文件大小为 2~7G 不等。需要注意的是,还有一些非 sra 格式的文件也会被下载下来,要和 sra 文件放在一起,后面做格式转换的时候程序会自动检索,如果没有放在一起,可能会报错。不过我们这里只关心 sra 文件。
4.8G SRR3182418.sra
3.0G SRR3182419.sra
3.1G SRR3182420.sra
3.1G SRR3182421.sra
......
3.1.2 文件重命名
## 首先提取 sra 文件的 id
cat SraRunTable.txt | grep "WXS" | cut -f 1 -d , > sra
## 然后提取 sra 文件对应的样本 id
cat SraRunTable.txt | grep "WXS" | cut -f 34 -d , | sed s/_1// >config
## 合并,结果如下:两列,第一列为 sra 文件的 id,第二列为重命名后的样本 id
paste sra config > sra2case.txt
cat sra2case.txt
# SRR3182418 case5_germline
# SRR3182419 case2_germline
# SRR3182420 case4_germline
# SRR3182421 case3_germline
# SRR3182422 case6_germline
# SRR3182423 case1_germline
# SRR3182425 case4_biorep_B_techrep
# SRR3182426 case4_biorep_C
# SRR3182427 case3_biorep_A
# SRR3182428 case3_biorep_B
# ......
## 用下面代码重命名
cat sra2case.txt | while read id
do
arr=(${id})
sample=${arr[0]}
case=${arr[1]}
mv ./0.sra/${sample}/${sample}.sra ./0.sra/${case}.sra
done
## 最后删掉没用的文件夹
rm -r 0.sra/SRR*
# nohup prefetch --option-file ../sra -t fasp -p -c -O . -X 50G > download_sra.log 2>&1 &
## vdb-validate
# touch validate.out
# ls SRR* | grep -v "vdb" | xargs vdb-validate - >> validate.out 2>&1
# cat validate.out | grep consistent | wc
处理后结果:
case1_biorep_A_techrep.sra
case1_biorep_B.sra
case1_biorep_C.sra
case1_germline.sra
case1_techrep.sra
......
3.1.3 格式转换 sra 转 fq
最开始将 sra 转成 fq 用到的命令是 fast-dump,但是速度很慢。新版本的 sra-tools ,可以使用更快的工具做格式转换 fasterq-dump。按照我的代码习惯,需要用到一个 config 配置文件,其实里面就是记录了样本的 id,后面我们会经常用到这个文件。
$ cat config
case1_biorep_A_techrep
case2_biorep_A
case3_biorep_A
case4_biorep_A
case5_biorep_A
case6_biorep_A_techrep
case1_biorep_B
case2_biorep_B_techrep
case3_biorep_B
case4_biorep_B_techrep
case5_biorep_B_techrep
case6_biorep_B
case1_biorep_C
case2_biorep_C
case3_biorep_C_techrep
case4_biorep_C
case5_biorep_C
case6_biorep_C
case1_techrep_2
case2_techrep_2
case3_techrep_2
case4_techrep_2
case5_techrep_2
case6_techrep_2
简单脚本处理数据:
cd ./0.sra
## fasterq-dump
cat config | while read id
do
time fasterq-dump -3 -e 16 ${id}.sra -O ../1.raw_fq --outfile ${id}.fastq >> ../1.raw_fq/${id}_sra2fq.log 2>&1
time pigz -p 10 -f ../1.raw_fq/${id}_1.fastq >../1.raw_fq/${id}_1.fastq.gz
time pigz -p 10 -f ../1.raw_fq/${id}_2.fastq >../1.raw_fq/${id}_2.fastq.gz
done
nohup bash sra2fq.sh &
解释一下上面的脚本,首先通过 fasterq-dump 将 sra 转成 fastq格式,-e 16 指的是调用了 16 个线程,输出到了 1.raw_fq 文件夹中,得到了原始的 fq 文件,因为是双端测序,所以每个样本会有 2 个 fastq 文件${id}_1.fastq 和 ${id}_2.fastq,对应 reads_1 和 reads_2,还有一个垃圾文件 ${id}_fastq。原始的 fastq 文件很大,所以通过 pigz 压缩命令调用了 -p 10 个线程来压缩,可以留意一下压缩后文件的大小。
5.8G case1_biorep_A_techrep_1.fastq.gz
5.8G case1_biorep_A_techrep_2.fastq.gz
4.9G case1_biorep_B_1.fastq.gz
4.9G case1_biorep_B_2.fastq.gz
6.1G case1_biorep_C_1.fastq.gz
6.1G case1_biorep_C_2.fastq.gz
2.4G case1_germline_1.fastq.gz
2.4G case1_germline_2.fastq.gz
......
3.2 下载fastq数据
3.2.1 安装 Aspera
cd ~/wes_cancer/biosoft
wget -c https://download.asperasoft.com/download/sw/connect/3.8.1/ibm-aspera-connect-3.8.1.161274-linux-g2.12-64.tar.gz
tar -zxvf ibm-aspera-connect-3.8.1.161274-linux-g2.12-64.tar.gz
bash ibm-aspera-connect-3.8.1.161274-linux-g2.12-64.sh
## 添加环境变量
echo 'export PATH=~/.aspera/connect/bin/:$PATH' >> ~/.bashrc
source ~/.bashrc
3.2.2 获取下载链接
通过 https://sra-explorer.info/?# 获取下载数据的 URL ,直接在搜索框输入要下载的数据 id 号,然后选择要下载的数据即可。
3.2.3 下载数据
$ cd ~/wes_cancer/project/1.raw_fq
$ cat >fq_download.sh
#!/usr/bin/env bash
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182418/SRR3182418.fastq.gz . && mv SRR3182418.fastq.gz SRR3182418_exome_sequencing_of_case5_germline.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182418/SRR3182418_1.fastq.gz . && mv SRR3182418_1.fastq.gz SRR3182418_exome_sequencing_of_case5_germline_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182418/SRR3182418_2.fastq.gz . && mv SRR3182418_2.fastq.gz SRR3182418_exome_sequencing_of_case5_germline_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182419/SRR3182419.fastq.gz . && mv SRR3182419.fastq.gz SRR3182419_exome_sequencing_of_case2_germline.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182419/SRR3182419_1.fastq.gz . && mv SRR3182419_1.fastq.gz SRR3182419_exome_sequencing_of_case2_germline_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182419/SRR3182419_2.fastq.gz . && mv SRR3182419_2.fastq.gz SRR3182419_exome_sequencing_of_case2_germline_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182421/SRR3182421.fastq.gz . && mv SRR3182421.fastq.gz SRR3182421_exome_sequencing_of_case3_germline.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182421/SRR3182421_1.fastq.gz . && mv SRR3182421_1.fastq.gz SRR3182421_exome_sequencing_of_case3_germline_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182421/SRR3182421_2.fastq.gz . && mv SRR3182421_2.fastq.gz SRR3182421_exome_sequencing_of_case3_germline_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182420/SRR3182420.fastq.gz . && mv SRR3182420.fastq.gz SRR3182420_exome_sequencing_of_case4_germline.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182420/SRR3182420_1.fastq.gz . && mv SRR3182420_1.fastq.gz SRR3182420_exome_sequencing_of_case4_germline_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182420/SRR3182420_2.fastq.gz . && mv SRR3182420_2.fastq.gz SRR3182420_exome_sequencing_of_case4_germline_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182422/SRR3182422.fastq.gz . && mv SRR3182422.fastq.gz SRR3182422_exome_sequencing_of_case6_germline.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182422/SRR3182422_1.fastq.gz . && mv SRR3182422_1.fastq.gz SRR3182422_exome_sequencing_of_case6_germline_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182422/SRR3182422_2.fastq.gz . && mv SRR3182422_2.fastq.gz SRR3182422_exome_sequencing_of_case6_germline_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182423/SRR3182423.fastq.gz . && mv SRR3182423.fastq.gz SRR3182423_exome_sequencing_of_case1_germline.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182423/SRR3182423_1.fastq.gz . && mv SRR3182423_1.fastq.gz SRR3182423_exome_sequencing_of_case1_germline_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182423/SRR3182423_2.fastq.gz . && mv SRR3182423_2.fastq.gz SRR3182423_exome_sequencing_of_case1_germline_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182424/SRR3182424.fastq.gz . && mv SRR3182424.fastq.gz SRR3182424_exome_sequencing_of_case4_biorep_A.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182424/SRR3182424_1.fastq.gz . && mv SRR3182424_1.fastq.gz SRR3182424_exome_sequencing_of_case4_biorep_A_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182424/SRR3182424_2.fastq.gz . && mv SRR3182424_2.fastq.gz SRR3182424_exome_sequencing_of_case4_biorep_A_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182426/SRR3182426.fastq.gz . && mv SRR3182426.fastq.gz SRR3182426_exome_sequencing_of_case4_biorep_C.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182426/SRR3182426_1.fastq.gz . && mv SRR3182426_1.fastq.gz SRR3182426_exome_sequencing_of_case4_biorep_C_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182426/SRR3182426_2.fastq.gz . && mv SRR3182426_2.fastq.gz SRR3182426_exome_sequencing_of_case4_biorep_C_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182425/SRR3182425.fastq.gz . && mv SRR3182425.fastq.gz SRR3182425_exome_sequencing_of_case4_biorep_B_techrep_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182425/SRR3182425_1.fastq.gz . && mv SRR3182425_1.fastq.gz SRR3182425_exome_sequencing_of_case4_biorep_B_techrep_1_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182425/SRR3182425_2.fastq.gz . && mv SRR3182425_2.fastq.gz SRR3182425_exome_sequencing_of_case4_biorep_B_techrep_1_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182429/SRR3182429.fastq.gz . && mv SRR3182429.fastq.gz SRR3182429_exome_sequencing_of_case3_biorep_C_techrep_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182429/SRR3182429_1.fastq.gz . && mv SRR3182429_1.fastq.gz SRR3182429_exome_sequencing_of_case3_biorep_C_techrep_1_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182429/SRR3182429_2.fastq.gz . && mv SRR3182429_2.fastq.gz SRR3182429_exome_sequencing_of_case3_biorep_C_techrep_1_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182428/SRR3182428.fastq.gz . && mv SRR3182428.fastq.gz SRR3182428_exome_sequencing_of_case3_biorep_B.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182428/SRR3182428_1.fastq.gz . && mv SRR3182428_1.fastq.gz SRR3182428_exome_sequencing_of_case3_biorep_B_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182428/SRR3182428_2.fastq.gz . && mv SRR3182428_2.fastq.gz SRR3182428_exome_sequencing_of_case3_biorep_B_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182430/SRR3182430.fastq.gz . && mv SRR3182430.fastq.gz SRR3182430_exome_sequencing_of_case6_biorep_A_techrep_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182430/SRR3182430_1.fastq.gz . && mv SRR3182430_1.fastq.gz SRR3182430_exome_sequencing_of_case6_biorep_A_techrep_1_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182430/SRR3182430_2.fastq.gz . && mv SRR3182430_2.fastq.gz SRR3182430_exome_sequencing_of_case6_biorep_A_techrep_1_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182427/SRR3182427.fastq.gz . && mv SRR3182427.fastq.gz SRR3182427_exome_sequencing_of_case3_biorep_A.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182427/SRR3182427_1.fastq.gz . && mv SRR3182427_1.fastq.gz SRR3182427_exome_sequencing_of_case3_biorep_A_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182427/SRR3182427_2.fastq.gz . && mv SRR3182427_2.fastq.gz SRR3182427_exome_sequencing_of_case3_biorep_A_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182431/SRR3182431.fastq.gz . && mv SRR3182431.fastq.gz SRR3182431_exome_sequencing_of_case6_biorep_B.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182431/SRR3182431_1.fastq.gz . && mv SRR3182431_1.fastq.gz SRR3182431_exome_sequencing_of_case6_biorep_B_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182431/SRR3182431_2.fastq.gz . && mv SRR3182431_2.fastq.gz SRR3182431_exome_sequencing_of_case6_biorep_B_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182432/SRR3182432.fastq.gz . && mv SRR3182432.fastq.gz SRR3182432_exome_sequencing_of_case6_biorep_C.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182432/SRR3182432_1.fastq.gz . && mv SRR3182432_1.fastq.gz SRR3182432_exome_sequencing_of_case6_biorep_C_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182432/SRR3182432_2.fastq.gz . && mv SRR3182432_2.fastq.gz SRR3182432_exome_sequencing_of_case6_biorep_C_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182433/SRR3182433.fastq.gz . && mv SRR3182433.fastq.gz SRR3182433_exome_sequencing_of_case1_biorep_A_techrep_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182433/SRR3182433_1.fastq.gz . && mv SRR3182433_1.fastq.gz SRR3182433_exome_sequencing_of_case1_biorep_A_techrep_1_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182433/SRR3182433_2.fastq.gz . && mv SRR3182433_2.fastq.gz SRR3182433_exome_sequencing_of_case1_biorep_A_techrep_1_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182436/SRR3182436.fastq.gz . && mv SRR3182436.fastq.gz SRR3182436_exome_sequencing_of_case2_biorep_A.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182436/SRR3182436_1.fastq.gz . && mv SRR3182436_1.fastq.gz SRR3182436_exome_sequencing_of_case2_biorep_A_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182436/SRR3182436_2.fastq.gz . && mv SRR3182436_2.fastq.gz SRR3182436_exome_sequencing_of_case2_biorep_A_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182434/SRR3182434.fastq.gz . && mv SRR3182434.fastq.gz SRR3182434_exome_sequencing_of_case1_biorep_B.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182434/SRR3182434_1.fastq.gz . && mv SRR3182434_1.fastq.gz SRR3182434_exome_sequencing_of_case1_biorep_B_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182434/SRR3182434_2.fastq.gz . && mv SRR3182434_2.fastq.gz SRR3182434_exome_sequencing_of_case1_biorep_B_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182437/SRR3182437.fastq.gz . && mv SRR3182437.fastq.gz SRR3182437_exome_sequencing_of_case2_biorep_B_techrep_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182437/SRR3182437_1.fastq.gz . && mv SRR3182437_1.fastq.gz SRR3182437_exome_sequencing_of_case2_biorep_B_techrep_1_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182437/SRR3182437_2.fastq.gz . && mv SRR3182437_2.fastq.gz SRR3182437_exome_sequencing_of_case2_biorep_B_techrep_1_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182438/SRR3182438.fastq.gz . && mv SRR3182438.fastq.gz SRR3182438_exome_sequencing_of_case2_biorep_C.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182438/SRR3182438_1.fastq.gz . && mv SRR3182438_1.fastq.gz SRR3182438_exome_sequencing_of_case2_biorep_C_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/008/SRR3182438/SRR3182438_2.fastq.gz . && mv SRR3182438_2.fastq.gz SRR3182438_exome_sequencing_of_case2_biorep_C_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182435/SRR3182435.fastq.gz . && mv SRR3182435.fastq.gz SRR3182435_exome_sequencing_of_case1_biorep_C.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182435/SRR3182435_1.fastq.gz . && mv SRR3182435_1.fastq.gz SRR3182435_exome_sequencing_of_case1_biorep_C_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182435/SRR3182435_2.fastq.gz . && mv SRR3182435_2.fastq.gz SRR3182435_exome_sequencing_of_case1_biorep_C_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182439/SRR3182439.fastq.gz . && mv SRR3182439.fastq.gz SRR3182439_exome_sequencing_of_case5_biorep_A.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182439/SRR3182439_1.fastq.gz . && mv SRR3182439_1.fastq.gz SRR3182439_exome_sequencing_of_case5_biorep_A_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/009/SRR3182439/SRR3182439_2.fastq.gz . && mv SRR3182439_2.fastq.gz SRR3182439_exome_sequencing_of_case5_biorep_A_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182441/SRR3182441.fastq.gz . && mv SRR3182441.fastq.gz SRR3182441_exome_sequencing_of_case4_techrep_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182441/SRR3182441_1.fastq.gz . && mv SRR3182441_1.fastq.gz SRR3182441_exome_sequencing_of_case4_techrep_2_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/001/SRR3182441/SRR3182441_2.fastq.gz . && mv SRR3182441_2.fastq.gz SRR3182441_exome_sequencing_of_case4_techrep_2_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182440/SRR3182440.fastq.gz . && mv SRR3182440.fastq.gz SRR3182440_exome_sequencing_of_case5_biorep_B_techrep_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182440/SRR3182440_1.fastq.gz . && mv SRR3182440_1.fastq.gz SRR3182440_exome_sequencing_of_case5_biorep_B_techrep_1_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/000/SRR3182440/SRR3182440_2.fastq.gz . && mv SRR3182440_2.fastq.gz SRR3182440_exome_sequencing_of_case5_biorep_B_techrep_1_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182442/SRR3182442.fastq.gz . && mv SRR3182442.fastq.gz SRR3182442_exome_sequencing_of_case3_techrep_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182442/SRR3182442_1.fastq.gz . && mv SRR3182442_1.fastq.gz SRR3182442_exome_sequencing_of_case3_techrep_2_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/002/SRR3182442/SRR3182442_2.fastq.gz . && mv SRR3182442_2.fastq.gz SRR3182442_exome_sequencing_of_case3_techrep_2_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182443/SRR3182443.fastq.gz . && mv SRR3182443.fastq.gz SRR3182443_exome_sequencing_of_case6_techrep_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182443/SRR3182443_1.fastq.gz . && mv SRR3182443_1.fastq.gz SRR3182443_exome_sequencing_of_case6_techrep_2_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/003/SRR3182443/SRR3182443_2.fastq.gz . && mv SRR3182443_2.fastq.gz SRR3182443_exome_sequencing_of_case6_techrep_2_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182444/SRR3182444.fastq.gz . && mv SRR3182444.fastq.gz SRR3182444_exome_sequencing_of_case1_techrep_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182444/SRR3182444_1.fastq.gz . && mv SRR3182444_1.fastq.gz SRR3182444_exome_sequencing_of_case1_techrep_2_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/004/SRR3182444/SRR3182444_2.fastq.gz . && mv SRR3182444_2.fastq.gz SRR3182444_exome_sequencing_of_case1_techrep_2_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182445/SRR3182445.fastq.gz . && mv SRR3182445.fastq.gz SRR3182445_exome_sequencing_of_case2_techrep_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182445/SRR3182445_1.fastq.gz . && mv SRR3182445_1.fastq.gz SRR3182445_exome_sequencing_of_case2_techrep_2_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/005/SRR3182445/SRR3182445_2.fastq.gz . && mv SRR3182445_2.fastq.gz SRR3182445_exome_sequencing_of_case2_techrep_2_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182446/SRR3182446.fastq.gz . && mv SRR3182446.fastq.gz SRR3182446_exome_sequencing_of_case5_techrep_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182446/SRR3182446_1.fastq.gz . && mv SRR3182446_1.fastq.gz SRR3182446_exome_sequencing_of_case5_techrep_2_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/006/SRR3182446/SRR3182446_2.fastq.gz . && mv SRR3182446_2.fastq.gz SRR3182446_exome_sequencing_of_case5_techrep_2_2.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182447/SRR3182447.fastq.gz . && mv SRR3182447.fastq.gz SRR3182447_exome_sequencing_of_case5_biorep_C.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182447/SRR3182447_1.fastq.gz . && mv SRR3182447_1.fastq.gz SRR3182447_exome_sequencing_of_case5_biorep_C_1.fastq.gz
ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/fastq/SRR318/007/SRR3182447/SRR3182447_2.fastq.gz . && mv SRR3182447_2.fastq.gz SRR3182447_exome_sequencing_of_case5_biorep_C_2.fastq.gz
运行脚本下载数据:
nohup bash fq_download.sh &
下载完成后,每个样本会有 3 个 fastq 文件,分别对应 *_1.fastq.gz 和 *_2.fastq.gz ,以及一个只有几百K没什么用的 fastq 文件,比如下面这个样本:
442K SRR3182418_exome_sequencing_of_case5_germline.fastq.gz
3.3G SRR3182418_exome_sequencing_of_case5_germline_1.fastq.gz
3.4G SRR3182418_exome_sequencing_of_case5_germline_2.fastq.gz
通常这个多余的 fastq 文件我都不关心,直接删掉,一个个删掉也有点麻烦,当然如果你的 Linux 基础比较好的话,是可以用命令来删除的:
find ./*gz -size 1M | xargs rm
## 结果
case1_biorep_A_techrep_1_1.fastq.gz
case1_biorep_A_techrep_1_2.fastq.gz
case1_biorep_B_1.fastq.gz
case1_biorep_B_2.fastq.gz
......
## 重命名
ls -l | grep '.gz' | cut -c 48- > config.txt
4 质控与去接头
从sra转格式后拿到的fq文件是测序原始的fq,记录了测序得到的reads的碱基序列和对应质量值,如case1_biorep_A_techrep_1.fastq.gz 的前8行,以每4行为单位,作为一条reads的完整记录:
@case1_biorep_A_techrep.1/1 1 length=76
AACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACC
+case1_biorep_A_techrep.1/1 1 length=76
A@?CDDABEDDDABDBBCAADBBCAADBCDABDCCC@BDBCCAADCCA@AECCCAAEDCDAAEDBDAAEDDA>=C=
@case1_biorep_A_techrep.2/1 2 length=76
GTTGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAG
+case1_biorep_A_techrep.2/1 2 length=76
@?=DDDA@AGEEA@@FCD??@FCD@AAGCC@@?GED@@@FCC=??FEE=@@DEE=??EDE=>;DDD@>>BBA><>>
其中第4行为质量值,由测序仪给出,根据Phred33(Phred64基本淘汰)可以换算成实际的质量值
但是每一个fq文件中都有上千万条reads,这样换算显然是不科学的。

4.1 fastqc 的使用
前面我们已经安装了fastqc软件,接下来我们就用这个软件来对fq文件进行统计可视化,看看reads的读长、质量等分布情况。首先也是要激活环境,然后进入工作目录
conda activate wes
cd ~/wes_cancer/project
同样的通过config文件来进行批量操作,脚本如下:
cat config | while read id
do
fastqc --outdir ./3.qc/raw_qc/ --threads 16 ./1.raw_fq/${id}*.fastq.gz >> ./3.qc/raw_qc/${id}_fastqc.log 2>&1
done
multiqc ./3.qc/raw_qc/*zip -o ./3.qc/raw_qc/multiqc
这里的参数 --threads16 指的是调用的服务器的16个线程来处理数据。
经过fastqc处理后,每个 fastq 文件会生成一个 html 文件和一个 zip 压缩文件,其中html文件是质控报告,可以下载到本地并用浏览器打开查看,而zip文件里面是 html 文件图表对应的数据。因为样本数较多,我们就用 multiqc 软件对所有样本的结果进行整合,方便一起查看。
4.2 质控和去接头
从上面的质控报告可以发现,有一些 reads 的质量值较差,或者测到了接头,这样的reads对于后续的分析是有一定的负面影响的,所以我们需要去除掉这些质量较差的reads或者切掉测到的接头,一般使用的软件是trim-galore,软件会自动检测已知的常用接头。
## trim_galore.sh
cat config | while read id
do
fq1=./1.raw_fq/${id}_1.fastq.gz
fq2=./1.raw_fq/${id}_2.fastq.gz
trim_galore --paired -q 28 --phred33 --length 30 --stringency 3 --gzip --cores 8 -o ./2.clean_fq $fq1 $fq2 >> ./2.clean_fq/${id}_trim.log 2>&1
done
nohup bash trim_galore.sh &
trim 处理后的生成文件名带有val标签,感兴趣可以比较 trim 前后的文件大小,但是这里我们还是再做一次 fastqc,通过质控报告来对比trim前后的结果
cat config | while read id
do
fastqc --outdir ./3.qc/clean_qc/ --threads 16 ./2.clean_fq/${id}*.fq.gz >> ./3.qc/clean_qc/${id}_fastqc.log 2>&1
done
multiqc ./3.qc/clean_qc/*zip -o ./3.qc/clean_qc/multiqc
5 比对与 bam 文件格式
5.1 参考基因组及索引
通过 trim 过滤后的到的 fq,需要比对到参考基因组上才能让这些数据有意义。前面我们已经下载了 人类参考基因组 hg38 版本,而且是从 GATK 数据库下载的,原文件大小为 800 多M,解压后为 3G 左右。同时也需要构建该参考基因组的 bwa 索引文件,即:
20K gatk_hg38.amb
3.0G gatk_hg38.bwt
1.5G gatk_hg38.sa
445K gatk_hg38.ann
768M gatk_hg38.pac
索引文件的作用类似于一本书的目录的作用,起到一个快速查找的功能。构建过程非常消耗时间和 服务器的资源,方法是:
conda activate wes
cd ~/wes_cancer/data
gunzip Homo_sapiens_assembly38.fasta.gz
time bwa index -a bwtsw -p gatk_hg38 ~/wes_cancer/data/Homo_sapiens_assembly38.fasta
cd ~/wes_cancer/project
构建过程中会产生很多信息:
[bwa_index] Pack FASTA... 14.70 sec
[bwa_index] Construct BWT for the packed sequence...
[BWTIncCreate] textLength=6434693834, availableWord=464768632
[BWTIncConstructFromPacked] 10 iterations done. 99999994 characters processed.
[BWTIncConstructFromPacked] 20 iterations done. 199999994 characters processed.
[BWTIncConstructFromPacked] 30 iterations done. 299999994 characters processed.
[BWTIncConstructFromPacked] 40 iterations done. 399999994 characters processed.
[BWTIncConstructFromPacked] 50 iterations done. 499999994 characters processed.
......
real 41m18.206s
user 40m47.192s
sys 2m30.302s
5.2 比对
有了索引文件之后,就可以开始比对,用到的比对软件是 bwa ,比对的 bwa.sh 脚本如下:
## bwa.sh
INDEX=~/wes_cancer/data/gatk_hg38
cat config | while read id
do
echo "start bwa for ${id}" `date`
fq1=./2.clean_fq/${id}_1_val_1.fq.gz
fq2=./2.clean_fq/${id}_2_val_2.fq.gz
bwa mem -M -t 16 -R "@RG\tID:${id}\tSM:${id}\tLB:WXS\tPL:Illumina" ${INDEX} ${fq1} ${fq2} | samtools sort -@ 10 -m 1G -o ./4.align/${id}.bam -
echo "end bwa for ${id}" `date`
done
bwa 比对后先产生 sam 文件,但是因为 sam 文件太大,占用空间,要转换成二进制的 bam 文件, 两者记录的信息是一样的。因此,通常都是把比对的结果通过管道符 | 传给 samtools 进行排序后 直接生存 bam 文件(注意最后还有一个中划线 “-”作为缓存占位符),排序的依据是 bam 文件 中 reads 比对到参考基因组的 染色体 和 坐标。生成的 bam 文件大小为 3 ~ 13G 不等。
13G case1_biorep_C.bam
11G case1_biorep_B.bam
12G case1_biorep_A_techrep.bam
......
3.4G case2_germline.bam
5.0G case4_germline.bam
7.4G case5_germline.bam
5.3 bam 文件格式
bam(或者是 sam,cram )文件,是比对后拿到的文件,sam 文件是纯文本,非常占用存储空 间,bam 是 sam 的二进制格式,cram 则是进一步压缩后的格式,这三者所记录的内容是一致的, 但是 bam 文件是最常用的。记录了每一条 reads 比对到参考基因组的结果,主要有 11 列比较重要 的信息(每一列以制表符分开):
| 列 | 列名 | 简介 | 举例 |
|---|---|---|---|
| 1 | QNAME | reads的名称 | case1_biorep_A_techrep.89335031 |
| 2 | FLAG | 由二进制数表示,每一个数字代表一种比对情 况,这里的值是符合情况的数字相加总和 | 5=1+22 |
| 3 | RNAME | 参考序列,一般是染色体 | chr1 |
| 4 | POS | 比对到染色体位置 | 42852401 |
| 5 | MAPQ | mapping质量 | 60 |
| 6 | CIGAR | MIDNSHP=XB这10个字符代表不同的含义,M比 对成功,I插入,D删除 | 76M3D |
| 7 | RNEXT | 配对reads比对到的参考序列(染色体) | chr11 |
| 8 | PNEXT | 配对reads比对到染色体的位置 | 96427099 |
| 9 | TLEN | 可以理解为测序是文库插入片段长度 | 0 |
| 10 | SEQ | 序列,fq文件的第二行 | GCTTC…CCAGC |
| 11 | QUAL | 质量值,fq文件第四行 | @@@?D…CA@DD |
更多的说明可以查看官方教程:https://samtools.github.io/hts-specs/SAMv1.pdf https://samtools.github.io/hts-specs/SAMv1.pdf
5.4 提取小 bam
由于 bam 文件特别大,下载到本地载入 IGV 非常耗资源,所以我们就提取一个小 bam 进行演示。 首先,要构建索引,提取小 bam,这里我们提取 17 号染色体的信息:chr17。再对小 bam 文件构 建索引,因为 IGV 要求 bam 文件需要带索引才能载入,以样本 case1_biorep_A_techrep 为 例。
samtools index case1_biorep_A_techrep.bam
samtools view -h case1_biorep_A_techrep.bam chr17 | samtools view -Sb - > small.bam
samtools index small.bam
对比一下大小,如果还觉得太大,上面提取的参数 chr17 可以改为类似 chr17:42850000- 42950000 ,即提取特定坐标的部分。
12G case1_biorep_A_techrep.bam
694M small.bam
5.5 载入 IGV
然后把小 bam 文件及其索引下载到本地,打开 IGV,加载 hg38 参考基因组,然后把 bam 文件拖 到 IGV 窗口中
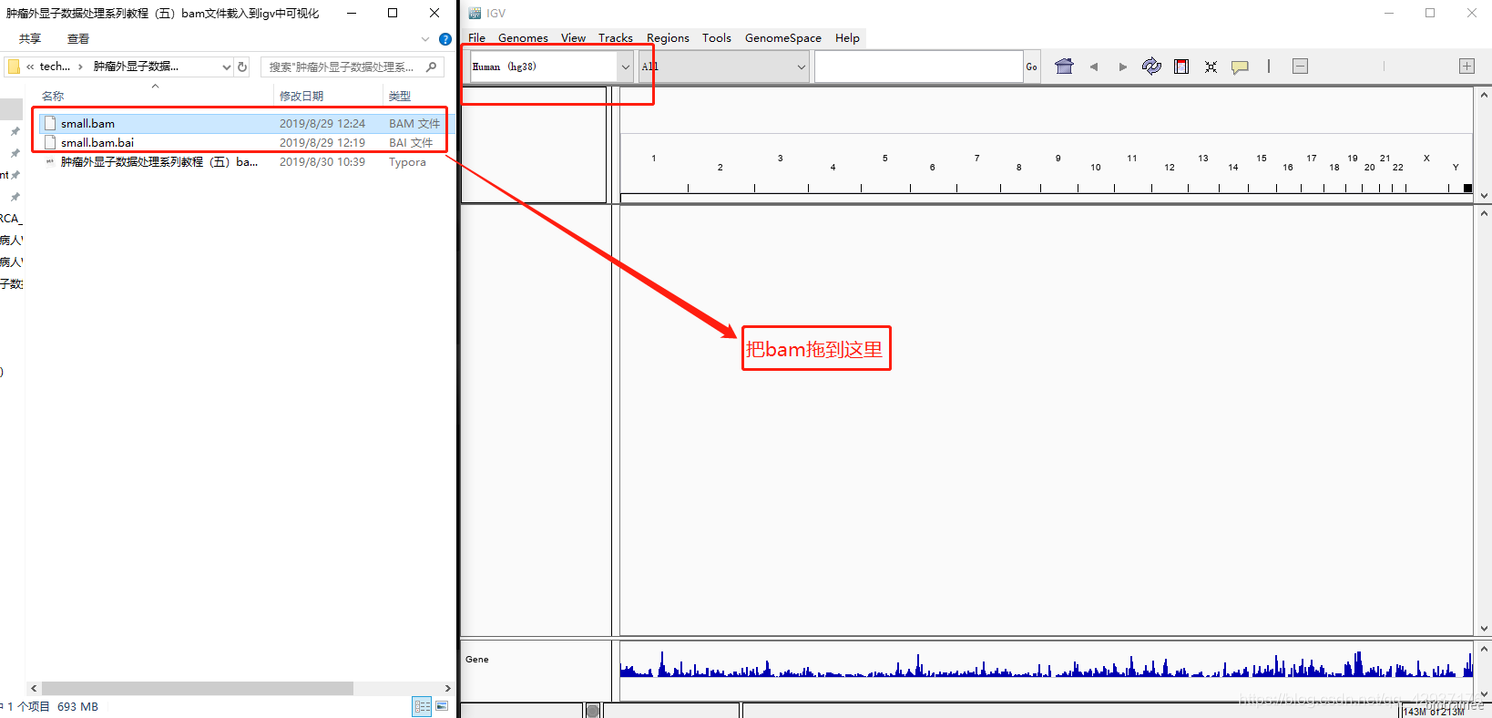
点击 17 和右上角的 +
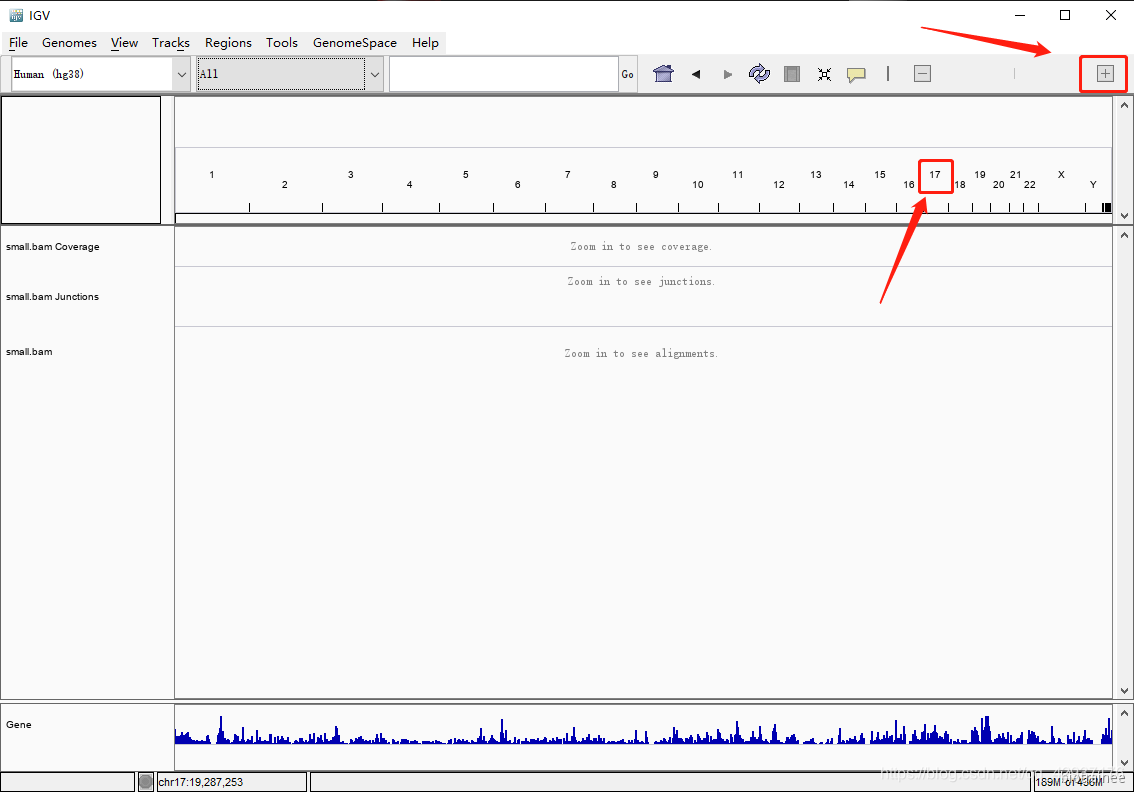
直到标尺显示的尺度小于 30kb( IGV 默认小于 30kb 才会显示 reads),就可以看到 reads 的信息
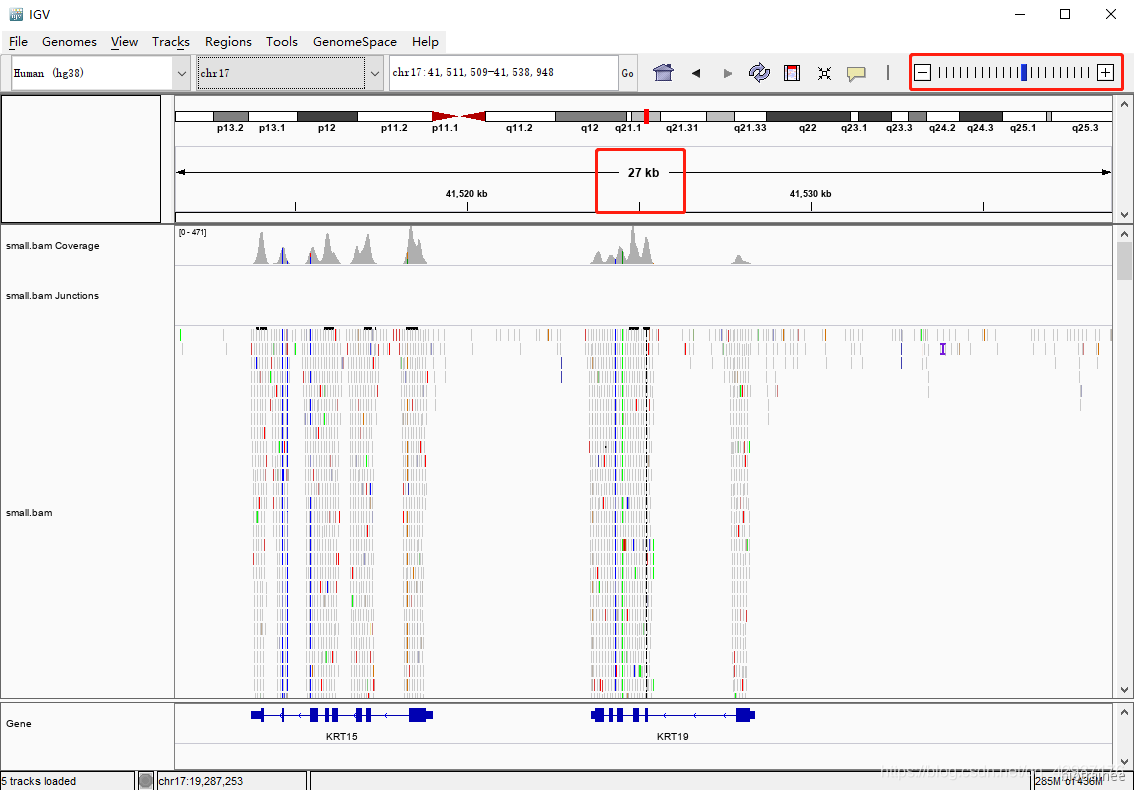
5.6 定位到感兴趣的位置
也可以在搜索框中输入感兴趣的位置或者基因,比如 chr17:42,850,644-42,853,784
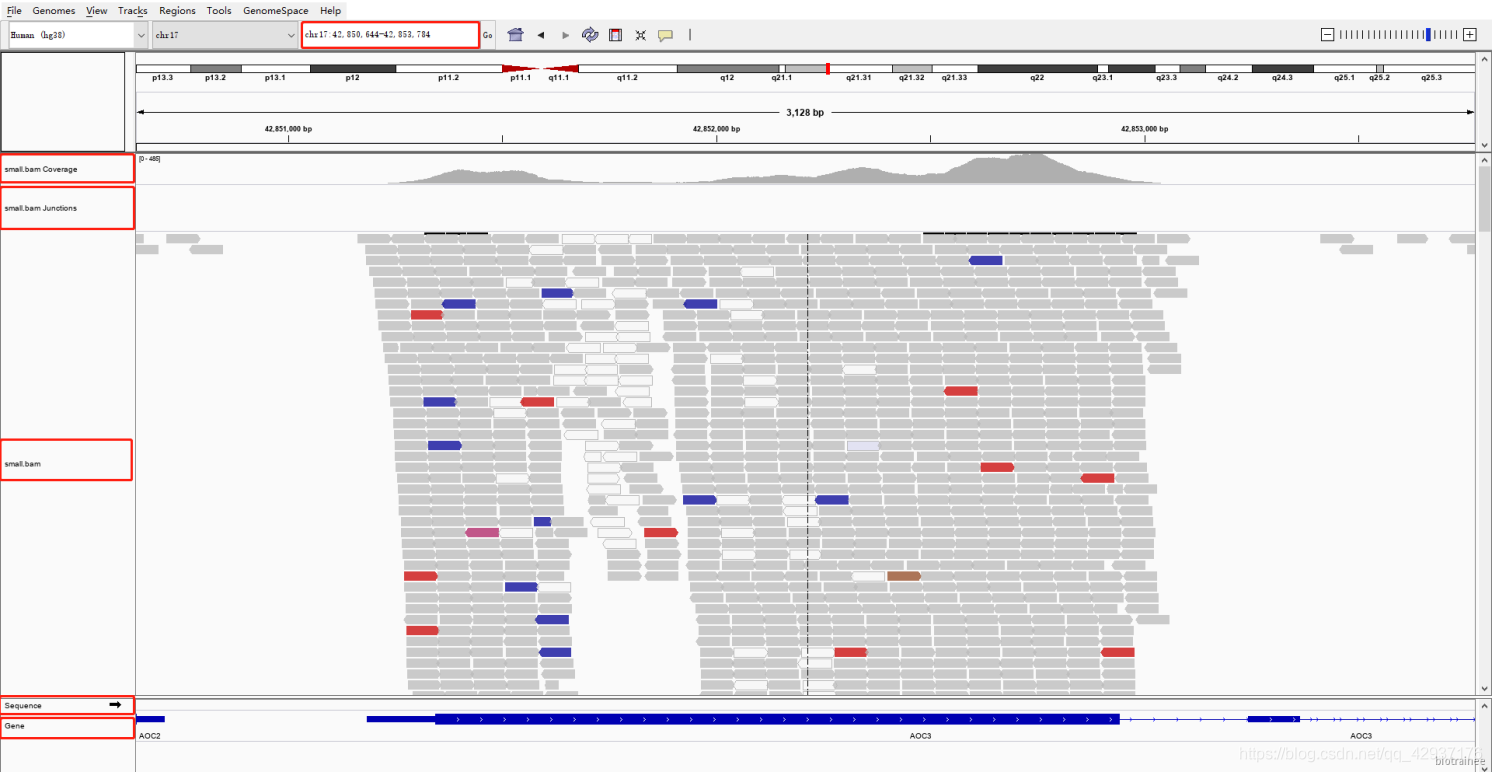
这里我们可以看到有5条 track:
- 第一条是 Coverage 即覆盖度,可以直观的看出染色体的每一处 reads 的覆盖情况,因为我们 是外显子捕获测序,所以极大部分的reads 都覆盖到了参考基因组的外显子区域及其侧翼区 域。值得注意的是在这一行开头有峰的高度范围,且默认是动态变化的。
- 第二条是junctions,一般用于转录组数据,可以看可变剪切,默认是不显示的,这里是我之前设置。
- 第三条是 bam文件中reads详细信息,每一块代表一条 reads,将鼠标放到某一条 reads 上, 就会呈现该 reads 的详细比对信息,与bam 文件的中信息相对应。而且可以通过鼠标右键,调出设置菜单,方便进一步探索。
- 第四条是sequence,参考基因组的序列碱基信息,当放大至一定程度时就会显示出来。
- 第五条是参考基因组注释信息,可以看到有基因、外显子、内含子、5’UTR、3’UTR 等,还可以 右键选择显示基因的各个转录本。
5.7 颜色代表的含义
可以看到,这些 reads 大多数是灰色的,部分是透明,少部分是红色、蓝色、棕色等等,不同的颜 色有不同的含义。
灰色:指的是比对成功,没有其他特别的含义
透明:指的是比对失败,对应的是 bam 文件中第 5 列,MAPQ 比对质量值,我们把鼠标放到透明 的 reads 上就可以看到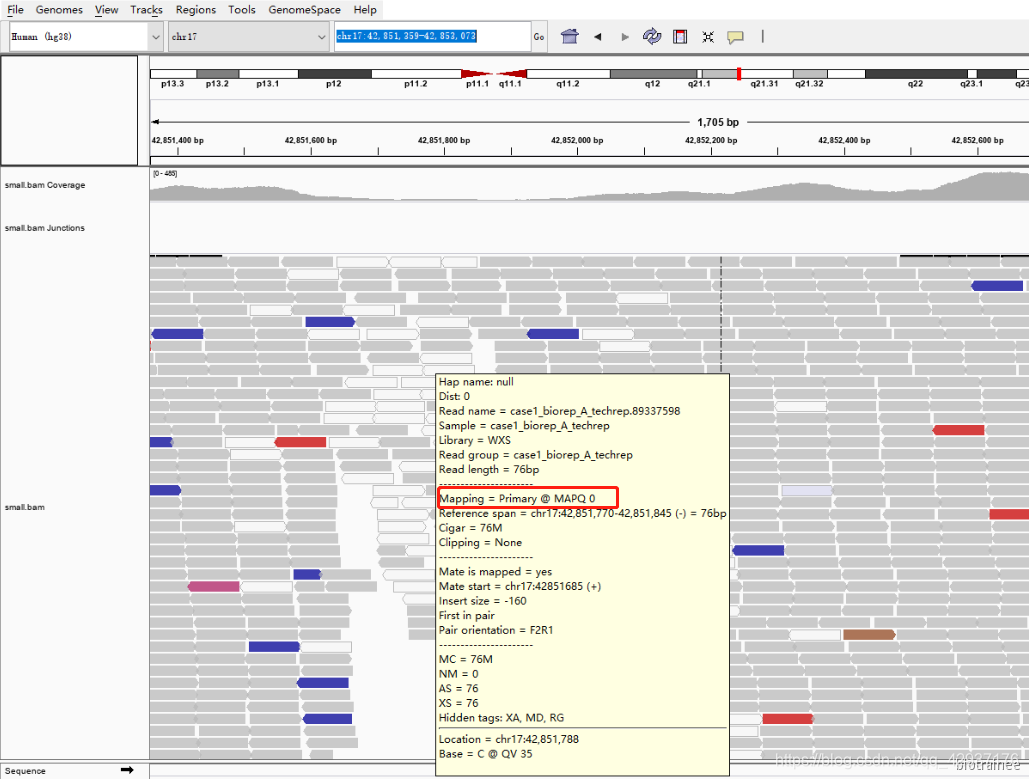
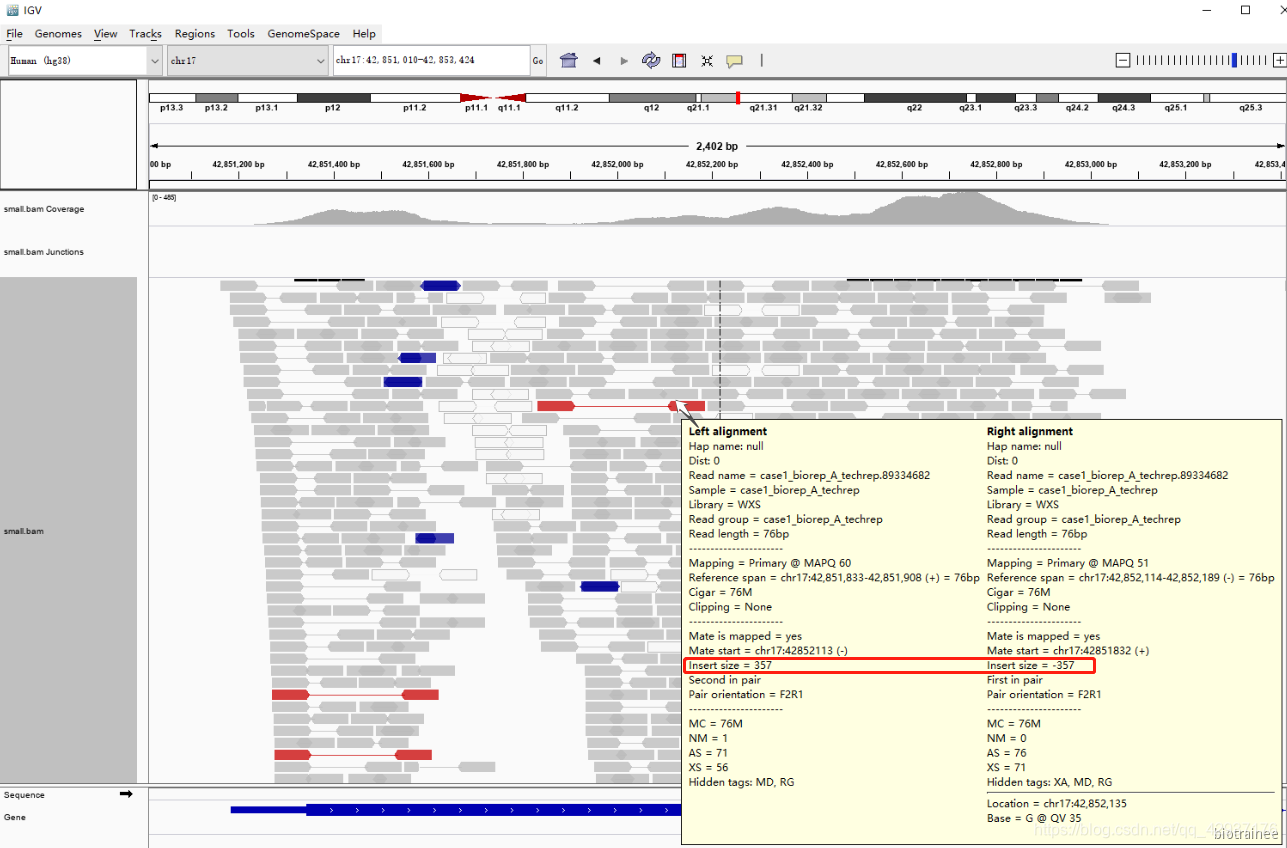
红色和蓝色: IGV 会根据配对的两条 reads 的距离,即 bam 的第 9 列 TLEN 测序是文库插入片段 长度,来判断是否存在染色体的结构变异。红色代表插入片段大于期望值,可能是染色体片段缺失 的证据,蓝色代表插入片段小于预期,可能是染色体插入的证据。我们可以通过右键选择 view as pairs 来进一步理解这个含义。蓝色的两条 reads 重叠,而红色的 reads 距离都比较大。
其他颜色:指的是这条 reads 所配对的另一条 reads 没有比对到同一条染色体,不同颜色代表不同 染色体,具体看下图:

比如下面棕色的 reads ,代表与之配对的reads比对到了 11 号染色体上了: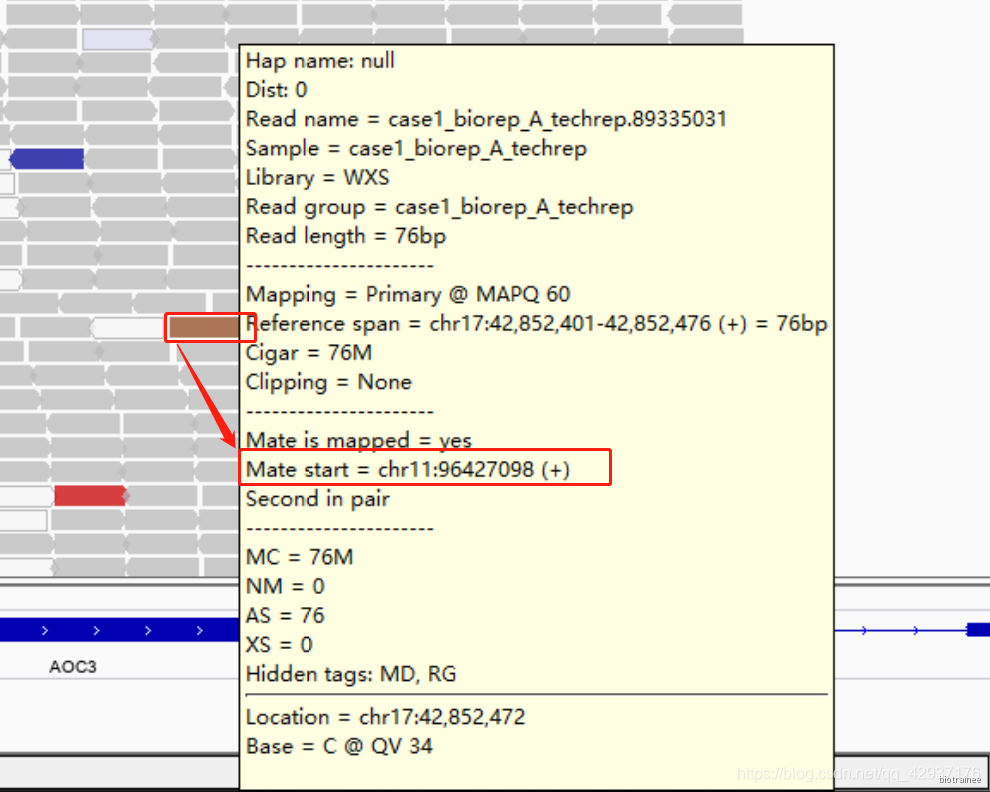
我们可以通过 bam 文件来检验一下,因为是 Pair-end,所以 id 号相同,可以 grep 89335031 :
关于颜色更多的介绍,请参考 IGV 官方文档:http://software.broadinstitute.org/software/igv/interpreting_insert_size
不同组学的测序策略不同,这里展示的是外显子组的 bam 文件,还有其他组学的 bam 文件也可以 在 IGV 中可视化,对此推荐大家看一下不同组学测序数据拿到的bam文件在 IGV 可视化结果的区别:[有参组学的NGS数据分析的异同点分享 ](https://mp.weixin.qq.com/s? __biz=MzAxMDkxODM1Ng==&mid=2247486104&idx=2&sn=ed79cf6c319184a75d359935643 9f08c&scene=21#wechat_redirect)
6 比对结果的质控
6.1 samtools stats
## stats.sh
cat config | while read id
do
bam=./4.align/${id}.bam
samtools stats -@ 16 --reference ~/wes_cancer/data/Homo_sapiens_assembly38.fasta ${bam} > ./4.align/stats/${id}.stat
plot-bamstats -p ./4.align/stats/${id} ./4.align/stats/${id}.stat
done
报bug:
gnuplot: error while loading shared libraries: libjpeg.so.8: cannot open shared object file: No such file or directory
解决方法:
sudo apt-get install libjpeg-turbo8
质控后生成html网页报告,可看到碱基分布信息和统计信息
Reads
total: 212,740,888
filtered: 0 (0.0%)
non-primary: 21,011
duplicated: 0 (0.0%)
mapped: 212,738,868 (100.0%)
zero MQ: 17,615,610 (8.3%)
avg read length: 74
Bases
total: 15,940,884,573
mapped: 15,935,687,200 (100.0%)
error rate: 0.20%
6.2 qualimap
用 qualimap 查看基因组覆盖深度等信息
cat config | while read id
do
qualimap bamqc --java-mem-size=10G -gff ~/wes_cancer/data/hg38.exon.bed -nr 100000 -nw 500 -nt 16 -bam ./4.align/${id}.bam -outdir ./4.align/qualimap/${id}
done
7 GATK流程和找变异
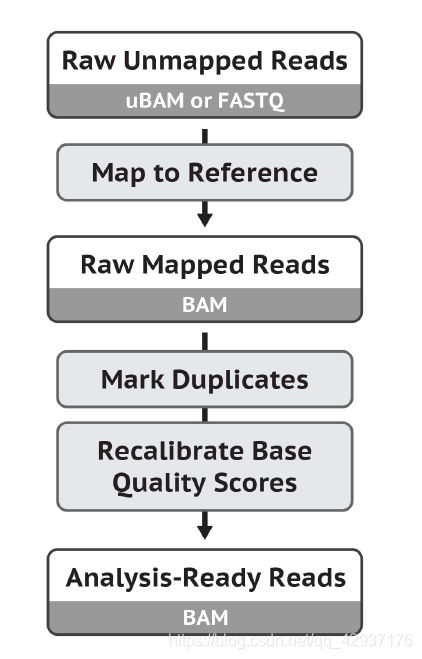
7.1 数据准备
···
dbsnp_146.hg38.vcf.gz
dbsnp_146.hg38.vcf.gz.tbi
Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi
Homo_sapiens_assembly38.fasta
Homo_sapiens_assembly38.fasta.gz
Homo_sapiens_assembly38.fasta.fai
Homo_sapiens_assembly38.dict
1000G_phase1.snps.high_confidence.hg38.vcf.gz
1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi
···
7.2 标记或去除重复序列
在拿到的测序结果中,有一部分序列是重复的,这是由于上机前需要进行PCR扩增,如果序列扩增 次数不同,就会导致PCR重复,重复序列在igv可视化的时候就可以明显看出来。比如我们前面提取 的small.bam在这个区域 chr17:43,719,044-43,720,613 的覆盖度就出现了断崖式的增长。
所以我们拿到比对后的bam文件之后,要进行标记重复序列甚至去除掉重复序列。
7.2.1 GATK MarkDuplicates 去重复
代码如下:
GATK=~/wes_cancer/biosoft/gatk-4.1.9.0/gatk
cat config | while read id
do
BAM=./4.align/${id}.bam
if [ ! -f ./5.gatk/ok.${id}_marked.status ]
then
echo "start MarkDuplicates for ${id}" `date`
$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" MarkDuplicates \
-I ${BAM} \
--REMOVE_DUPLICATES=true \
-O ./5.gatk/${id}_marked.bam \
-M ./5.gatk/${id}.metrics \
1>./5.gatk/${id}_log.mark 2>&1
if [ $? -eq 0 ]
then
touch ./5.gatk/ok.${id}_marked.status
fi
echo "end MarkDuplicates for ${id}" `date`
samtools index -@ 16 -m 4G -b ./5.gatk/${id}_marked.bam ./5.gatk/${id}_marked.bai
fi
done
解释一下上面的代码:首先把 config 文件做好,也就是我们的样本名,在这里作为一个配置文件, 在前面我们也一直用它,里面记录的内容是:
$ samtools view ./4.align/case1_biorep_A_techrep.bam |wc -l
212761899
$ samtools view ./5.gatk/case1_biorep_A_techrep_marked.bam |wc -l
192699040
进入到 while read id;do…;done 循环后,每 read 读取一行为一个 id,也就是一个样本名, 对于每一个样本,再赋值 bam 文件的具体路径,命名为 BAM 。
接下来就是一个 if 判断语句,判断是否已经存在生成的 ./5.gatk/ok.
i
d
m
a
r
k
e
d
.
s
t
a
t
u
s
∗
∗
文
件
,
如
果
存
在
,
就
不
再
执
行
,
如
果
不
存
在
就
继
续
往
下
走
。
需
要
注
意
的
是
,
如
果
在
s
h
e
l
l
命
令
执
行
后
,
会
有
一
个
返
回
值
∗
∗
{id}_marked.status** 文件,如果存在,就不再执行,如果不存在就继续往下走。需要注意的是,如果在 shell 命令执行后, 会有一个返回值 **
idmarked.status∗∗文件,如果存在,就不再执行,如果不存在就继续往下走。需要注意的是,如果在shell命令执行后,会有一个返回值∗∗? ,如果返回值为 0 ,只上一步命令成功执行,否则执行失败。在这里,如果命 令成功执行,我就创建一个空文件 ok.${id}_marked.status 。这样写控制语句的原因,是为了避免重复提交代码。如果多个样本批量跑,跑到一半的时候任务挂 了,比如服务器突然停电了,那后续再次提交命令的时候,已经完成的样本就不会被再次执行,我 们要做的就是重新提交这个脚本而已。紧接着就是 echo start…date 这是为了记录时间,和后面的 echo end…date 相对应,这样我们可以知道跑完一个样本需要的时间,也可以在命令前加 上一个 time 命令,也可以达到类似的效果。中间部分就是主要的命令:
-Xmx20G -Djava.io.tmpdir=./ :设置调用的内存大小为 20G
-I $BAM :指定输入文件
–REMOVE_DUPLICATES=true :删除掉重复序列,如果不加这个参数,就只是标记重复序列而不会删除。
-O ${id}_marked.bam :输出文件
*1>KaTeX parse error: Expected 'EOF', got '&' at position 17: …id}_log.mark 2>&̲1**:这里的1和2是捕获正确…{id}_log.mark
最后是用samtools对生成的bam文件构建索引,这是后续步骤的要求。
去重复其他方法:
samtools markdup -r test.bam markdup.bam
java -jar picard.jar MarkDuplicate REMOVE_DUPLICATES I=test.bam O=picard1.bam M=picard1.txt
简单查看一下删除重复序列的结果,去除 Duplicates 之前的总 reads 数为 212761899,删掉 Duplicates 之后剩下 192699040 了:
$ samtools view ./4.align/case1_biorep_A_techrep.bam |wc -l
212761899
$ samtools view ./5.gatk/case1_biorep_A_techrep_marked.bam |wc -l
192699040
7.2.2 picard.jar去重复
Picard 官网及教程:https://broadinstitute.github.io/picard/
下载:https://github.com/broadinstitute/picard/releases/latest
# picard.jar去重复
cat config | while read id
do
java -jar ~/wes_cancer/biosoft/picard.jar MarkDuplicates REMOVE_DUPLICATES=true I=../4.align/${id}.bam O=${id}_marked.bam M=${id}_metrics.txt
done
7.3 碱基质量重校正BQSR
因为对于 WGS 或者 WES 的测序数据,我们最关心的问题就是变异检测了,而变异检测和碱基质量 值密切相关。而碱基的质量值有测序仪和测序系统产生,机器带来的系统误差和噪声是不可避免 的。因此,找变异之前,需要进行碱基质量重校正BQSR(Base Quality Score Recalibration)。用 到了GATK的两个工具 BaseRecalibrator 和 ApplyBQSR ,代码如下:
GATK=~/wes_cancer/biosoft/gatk-4.1.9.0/gatk
snp=~/wes_cancer/data/dbsnp_146.hg38.vcf.gz
indel=~/wes_cancer/data/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
ref=~/wes_cancer/data/Homo_sapiens_assembly38.fasta
cat config | while read id
do
if [ ! -f ./5.gatk/${id}_bqsr.bam ]
then
echo "start BQSR for ${id}" `date`
$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" BaseRecalibrator \
-R $ref \
-I ./5.gatk/${id}_marked.bam \
--known-sites ${snp} \
--known-sites ${indel} \
-O ./5.gatk/${id}_recal.table \
1>./5.gatk/${id}_log.recal 2>&1
$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" ApplyBQSR \
-R $ref \
-I ./5.gatk/${id}_marked.bam \
-bqsr ./5.gatk/${id}_recal.table \
-O ./5.gatk/${id}_bqsr.bam \
1>./5.gatk/${id}_log.ApplyBQSR 2>&1
echo "end BQSR for ${id}" `date`
fi
done
关于上面的两个工具的解释:
第一个,BaseRecalibrator,这里计算出了所有需要进行重校正的reads和特征值,然后把这些信息 输出为一份校准表文件(recal.table)。 --known-sites 参数需要输入已知且可靠的变异位点,可 以有多个。因为人与人之间基因组的差异其实是很小的,约 0.1%,那么在一个群体中发现的已知变 异,在某个人身上也很可能是同样存在的。所以,我们首先排除掉所有的已知变异位点,然后计算 每个(报告出来的)质量值下面有多少个碱基在比对之后与参考基因组上的碱基是不同的,这些不 同碱基就被我们认为是错误的碱基,它们的数目比例反映的就是真实的碱基错误率。
第二个,ApplyBQSR,这一步利用第一步得到的校准表文件(recal.table)重新调整原来BAM文件 中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
校正前后的碱基质量值:
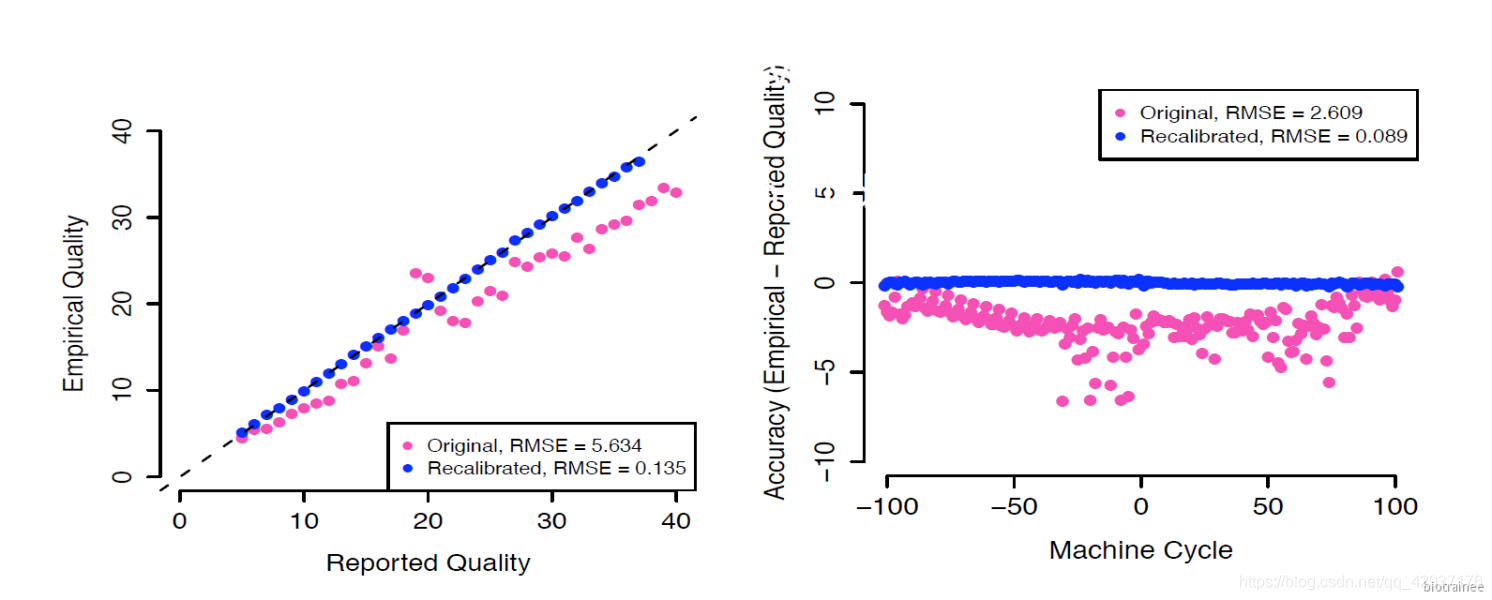
7.4 单样本找 Germline SNPS + INDELs
我们通常说的突变,可以分为种系突变 Germline mutations(又称生殖突变)和体细胞突变 Somatic mutations。前者是指在生殖细胞中发生的任何可检测、可遗传的突变。在生殖细胞系以外 的细胞中发生的突变称为体细胞突变,也称作获得性突变,是不可遗传的。使用 GATK 的流程进行 Germline mutations,主要用到的工具是 HaplotypeCaller,流程示意图如下(最后得到的 vcf 文 件应该还有走 VQSR 进行过滤):

GATK=~/wes_cancer/biosoft/gatk-4.1.9.0/gatk
snp=~/wes_cancer/data/dbsnp_146.hg38.vcf.gz
indel=~/wes_cancer/data/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
ref=~/wes_cancer/data/Homo_sapiens_assembly38.fasta
bed=~/wes_cancer/data/hg38.exon.bed
cat config | while read id
do
echo "start HC for ${id}" `date`
$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" HaplotypeCaller -ERC GVCF \
-R ${ref} \
-I ./5.gatk/${id}_bqsr.bam \
--dbsnp ${snp} \
-L ${bed} \
-O ./5.gatk/${id}_raw.vcf \
1>./5.gatk/${id}_log.HC 2>&1
echo "end HC for ${id}" `date`
done
cd ./5.gatk/gvcf
for chr in chr{1..22} chrX chrY chrM
do
time $GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" GenomicsDBImport \
-R ${ref} \
$(ls ./*raw.vcf | awk '{print "-V "$0" "}') \
-L ${chr} \
--genomicsdb-workspace-path gvcfs_${chr}.db
time $GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" GenotypeGVCFs \
-R ${ref} \
-V gendb://gvcfs_${chr}.db \
-O gvcfs_${chr}.vcf
done
$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" GatherVcfs \
$(for i in {1..22} X Y M;do echo "-I gvcfs_chr${i}.vcf" ;done) \
-O merge.vcf
7.5 mutect 找 Somatic mutations
前面单样本走 GATK HaplotypeCaller 流程得到 vcf 文件主要包含了 germline mutations 信息。但 是我们这里分析的是肿瘤样本,包含成对的 tumor 和 normal 样本,更加关心的是 tumor 样本中所 包含的 somatic mutations 信息。所以我们应该走 GATK 的 somatic mutations 流程:
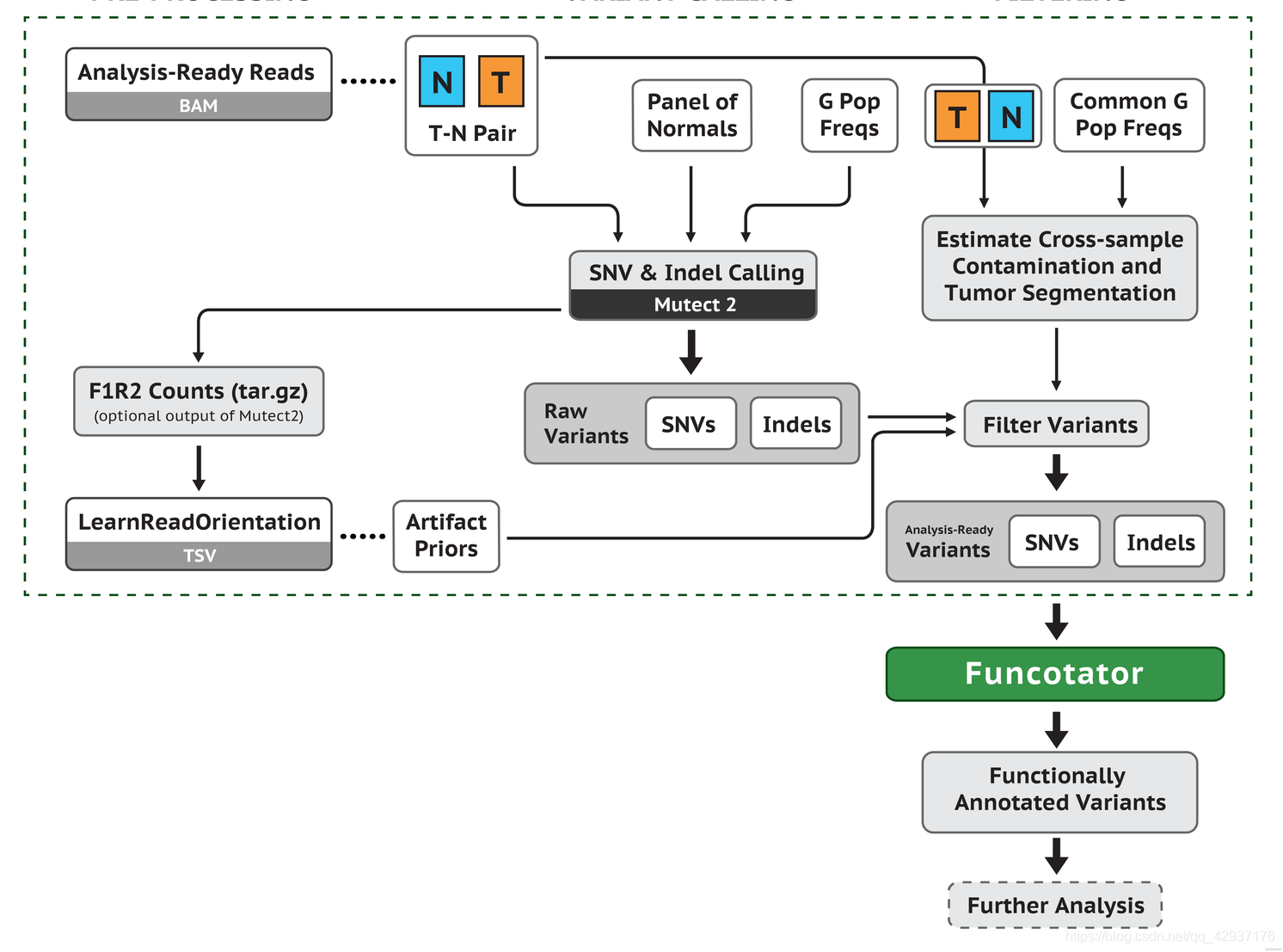
我们这里有成对的 normal 和 tumor样本,需要把配置文件 config2 做成两列,如下:
$ cat config2
case1_germline case1_biorep_A_techrep
case2_germline case2_biorep_A
case3_germline case3_biorep_A
......
case4_germline case4_techrep_2
case5_germline case5_techrep_2
case6_germline case6_techrep_2
运行脚本:
## mutect.sh
GATK=~/wes_cancer/biosoft/gatk-4.1.9.0/gatk
ref=~/wes_cancer/data/Homo_sapiens_assembly38.fasta
bed=~/wes_cancer/data/hg38.exon.bed
cat config2 | while read id
do
arr=(${id})
sample=${arr[1]}
T=./5.gatk/${arr[1]}_bqsr.bam
N=./5.gatk/${arr[0]}_bqsr.bam
echo "start Mutect2 for ${id}" `date`
$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" Mutect2 -R ${ref} \
-I ${T} -tumor $(basename "$T" _bqsr.bam) \
-I ${N} -normal $(basename "$N" _bqsr.bam) \
-L ${bed} \
-O ./6.mutect/${sample}_mutect2.vcf
$GATK FilterMutectCalls \
-R ${ref} \
-V ./6.mutect/${sample}_mutect2.vcf \
-O ./6.mutect/${sample}_somatic.vcf
echo "end Mutect2 for ${id}" `date`
cat ./6.mutect/${sample}_somatic.vcf | perl -alne '{if(/^#/){print}else{next unless $F[6] eq "PASS";next if $F[0] =~/_/;print } }' > ./6.mutect/${sample}_filter.vcf
done
关于上面代码的解释:首先是 Mutect2 ,主要是根据对正常-肿瘤样本进行位点比较寻找体细胞突 变 somatic mutations 。如果没有正常样本,也可以使用mutect2软件,流程需要进行修改,而 且结果的假阳性会很高。我们这里输入了两个配对的 T-N 样本 -I ${T} -tumor ( b a s e n a m e " (basename " (basename"T" _bqsr.bam) 和 -I ${N} -normal ( b a s e n a m e " (basename " (basename"N" _bqsr.bam) 。这样我们就拿到了 每个 tumor 样本中的突变位点, ${sample}_mutect2.vcf 。但是这还不够,还要通过 FilterMutectCalls 过滤,过滤后就是可信度较高的somatic突变位点会被打上 PASS 标签,就是 ${sample}_somatic.vcf 。最后我们再用一个简单的脚本把打上 PASS 标签的位点提取出来,得 到了 ${sample}_filter.vcf ,如:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT case1_biorep>
chr1 6146376 . G T . PASS DP=184;ECNT=1;NLOD=22.56;N_A>
chr1 6461445 . G T . PASS DP=21;ECNT=1;NLOD=2.70;N_ART>
chr1 31756671 . C A . PASS DP=172;ECNT=1;NLOD=9>
chr1 32672798 . A T . PASS DP=448;ECNT=1;NLOD=2>
chr1 39441098 . G T . PASS DP=411;ECNT=1;NLOD=3>
......
到这里拿到了最后的 vcf 文件,我们就可以进一步分析,而GATK的流程也到这里差不多结束,希望 你也能学会这个分析流程。















 275
275











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









