







小罗碎碎念
今天推荐6篇文章,都是AI病理领域最新发表的高分文章。

交流群
欢迎大家来到【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
公众号发表的文章会分享到群里,学术会议和比赛也会不定期分享,欢迎组队参与和线下交流!!
最后再介绍一下小罗自己:课题组位于广州,专业方向是医学人工智能,研究内容主要是病理组学,目前已借助自媒体与北大、北航、南方医等院校的课题组建立了联系,欢迎更多的人加入我们的队伍!!

一、突破传统:弱监督学习在有丝分裂检测中的革命性应用

文献概述
这篇文章是关于在乳腺组织病理图像中自动检测有丝分裂细胞的研究。作者提出了一种新颖的弱监督下的有丝分裂检测模型,该模型由一个候选提议网络和一个验证网络组成。候选提议网络基于图像块学习,目的是将有丝分裂细胞及其模拟物与背景分离,从而显著减少筛选过程中的漏检。然后,这些候选结果被送入验证网络进行有丝分裂细化。
双阶段有丝分裂检测模型的整体流程,包括两个主要部分:候选提议网络和验证网络。

-
候选提议网络 (Candidate Proposal Network):
- 这个网络的目的是筛选出可能的有丝分裂细胞目标。
- 它通过从背景中识别出候选对象,这些候选对象包括真正的有丝分裂细胞以及可能的模拟物(即外观相似但并非真正的有丝分裂细胞)。
- 这一步骤是初步的,旨在生成一个包含潜在有丝分裂事件的候选列表。
-
验证网络 (Verification Network):
- 经过候选提议网络筛选出的候选对象随后被送入验证网络。
- 验证网络的任务是对这些候选对象进行进一步的分类细化,以提高检测的准确性。
- 它采用基于径向基函数(RBF)的子分类方案来处理有丝分裂细胞内部的高变异性问题,以及区分外观相似的非有丝分裂细胞(即模拟物)。
整个流程强调了两个阶段的重要性:首先是生成候选对象,其次是对这些候选对象进行精确分类。这种双阶段方法有助于减少漏检(错过真正的有丝分裂事件)和误检(错误地将非有丝分裂细胞识别为有丝分裂细胞),从而提高有丝分裂检测的整体性能。
验证网络采用了基于径向基函数(RBF)的子分类方案,以处理有丝分裂细胞内部多样性高和外观相似的模拟物问题。通过联合训练分类和聚类,利用RBF中心来定义包含具有相似属性的有丝分裂细胞的子类别,并捕获具有代表性的RBF中心位置。由于子类别内部的变异较小,子类别级别的局部特征空间可以更好地表征某种类型的有丝分裂图像,并能提供更好的相似性度量,以区分有丝分裂细胞和非有丝分裂细胞。
实验结果表明,这种子分类方案有助于提高有丝分裂检测的性能,并在公开可用的有丝分裂数据集上取得了最先进的结果,仅使用了弱标签。作者还讨论了未来工作的方向,包括探索如何将领域表示引入深度RBF与子分类架构中,以提高有丝分裂领域的泛化能力。
在 MITOSIS2014 和 MIDOG 数据集上应用基于径向基函数(RBF)的子分类模型进行有丝分裂检测的一些示例
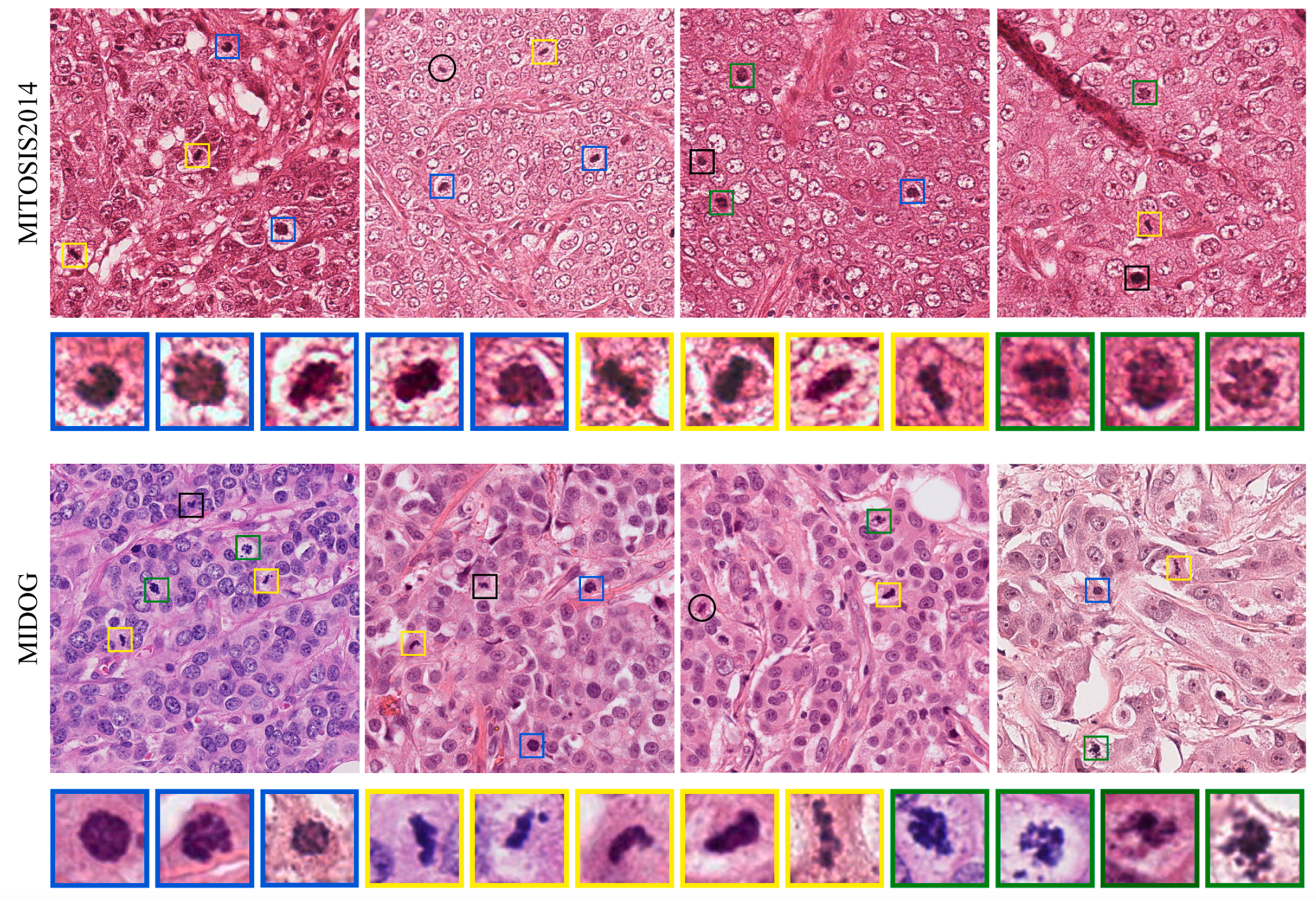
在这个图中,不同的颜色和标记用来表示模型的不同检测结果:
-
黄色、绿色和蓝色框:这些都是
真阳性(True Positives, TP),意味着模型正确地识别出了有丝分裂细胞。这些颜色的框分别对应于由RBF子分类生成的三个不同的子类别。RBF子分类有助于根据有丝分裂细胞的特征将它们分为不同的组,在这个例子中,每个颜色代表一个特定的子类别。 -
黑色框:这些是
假阳性(False Positives, FP),意味着模型错误地将非有丝分裂细胞识别为有丝分裂细胞。这可能是因为这些非有丝分裂细胞在视觉上与有丝分裂细胞非常相似,导致模型发生了误判。 -
黑色圆圈:这些表示
假阴性(False Negatives, FN),即模型未能检测到实际存在的有丝分裂细胞。这些有丝分裂细胞可能不够明显或太小,以至于模型无法检测到。 -
底部的放大视图:展示了真阳性检测结果的放大细节,以便更清楚地看到模型正确识别的有丝分裂细胞的具体特征。
总的来说,上图旨在展示RBF子分类模型在有丝分裂检测任务中的性能,包括它在区分真正的有丝分裂细胞以及抑制假阳性和假阴性方面的能力。通过子分类,模型能够更细致地分析和识别有丝分裂细胞,从而提高检测的准确性。
重点关注
使用颜色归一化方法进行颜色转移的一些示例

具体来说,这是从目标图像到源图像的颜色转移。这个方法是由 Vahadane 等人在 2016 年提出的,旨在通过颜色归一化处理减少不同扫描仪和染色方法导致的图像颜色表现的差异。
颜色归一化是一种预处理步骤,它可以帮助改善深度学习模型在处理来自不同来源的图像时的泛化能力。在组织病理学图像分析中,由于不同的扫描仪和染色技术,图像之间的颜色表现可能会有显著差异。这种差异可能会影响后续图像分析算法的性能,尤其是那些依赖于颜色信息的算法。
颜色转移的目的是将一组图像的颜色分布调整为目标图像的颜色分布,从而使得不同图像之间的颜色表现更加一致。这样,模型在训练时可以更加关注于图像中的结构信息,而不是由于颜色差异带来的干扰。
上图展示了几组图像对比,其中包括:
- 源图像:未经颜色归一化的原始图像,可能存在颜色偏差。
- 目标图像:颜色分布被用作标准的图像,通常选择颜色表现比较均衡和典型的图像。
- 归一化后的图像:源图像经过颜色转移处理后,其颜色分布与目标图像更加接近。
通过这种方式,颜色归一化有助于减少不同扫描仪和染色条件对图像分析算法性能的影响,从而提高算法的稳定性和可靠性。这对于在多中心临床试验中分析病理图像尤为重要,因为图像数据可能来自不同的医疗设备和实验室条件。
二、癌症研究的未来: 人工智能如何重塑我们对疾病的理解

文献概述
这篇文章是《Nature Reviews Cancer》杂志上的一篇综述,名为"A guide to artificial intelligence for cancer researchers"。文章的主要目的是为癌症研究人员提供关于人工智能(AI)的实用指南。
以下是对文章内容的概括:
-
AI的普及:AI已经从专业资源转变为癌症研究人员易于获取的工具。基于AI的工具不仅可以提高日常工作效率,还可以从现有数据中提取隐藏信息,从而促进新的科学发现。
-
AI工具的基本素养:对于每个癌症研究人员来说,建立对这些工具的基本理解是有用的。传统生物科学背景的研究人员可以通过现成的软件使用AI工具,而那些对计算更感兴趣的研究人员可以开发自己的AI软件流程。
-
AI的一般原则:文章传达了AI在图像分析、自然语言处理和药物发现中的应用的一般原则。
-
深度学习:作为AI的一种常见类型,深度学习可以有效地处理图像和文本等非结构化数据。介绍了
监督学习、无监督学习、自监督学习和强化学习等深度学习的不同类型。 -
神经网络架构:讨论了
卷积神经网络(CNNs)和长短期记忆(LSTM)网络等深度学习模型,以及变换器神经网络(transformers)在图像和语言处理任务中的应用。 -
AI在癌症研究中的应用:深度学习在癌症研究中有广泛的应用,包括通过用户友好的工具分析显微镜图像和基因组信息,以及在药物发现中筛选潜在化合物。
-
生物医学图像分析:深度学习的发展扩大了基于计算机的图像分析的应用范围,特别是在组织病理学图像分析方面。
-
AI在放射学中的应用:介绍了AI如何通过计算机视觉模型增强放射学图像的分析能力,提高诊断准确性和个性化治疗计划。
-
AI在语言处理中的应用:
大型语言模型(LLMs)的出现显著提高了计算机处理自然语言的能力,这些模型可以执行文本的重构、总结或翻译,并可以合成新文本。 -
结论:文章强调了将AI应用于癌症研究的潜力,尤其是在挖掘肿瘤学中现实世界数据(RWD)的潜力方面。同时指出了实现AI在精准肿瘤学中应用的潜力所面临的挑战,包括数据整合、开发可解释和透明的AI模型、建立数据共享和模型验证的标准等。
重点关注
人工智能(AI)在癌症研究中的工作流程

这个图表分为四个部分(a、b、c、d),每个部分描述了不同的AI应用方法和研究领域。
a部分:介绍了深度学习如何连接基础研究、转化研究和临床研究。深度学习是AI的一个子集,它通过使用多层神经网络来学习和提取原始数据中的复杂特征和模式。
b部分:描述了基于AI的图像处理中常用的三种方法之一:标准图像分析任务。这些任务包括细胞计数或分割感兴趣区域(ROIs)。这类任务通常需要快速解决方案,并且有现成的工具可以使用。例如,ilastik适用于亚细胞分析,QuPath和3D Slicer分别常用于组织水平和全身成像。Fiji、ImageJ和CellProfiler是多功能工具,能够处理不同尺度的广泛图像分析任务。这些工具的选择性列表在补充表1中展示。
c部分:介绍了另一种图像分析任务类别,这些任务需要在大量数据集上进行迭代训练和微调。这些工具通常非常特定于任务,旨在解决一个特定问题。构建、训练和验证这些流程需要编程技能。
d部分:讨论了图像分析的未来趋势,即基础模型的发展。这些模型在广泛、异构的数据集上进行预训练,并且可以在众多任务上进行微调。在零样本学习(zero-shot learning)中,模型直接应用于测试集,而不需要任何特定任务的训练。而在少样本学习(few-shot learning)中,模型会提供少量标记样本以适应特定任务。
计算机视觉中人工智能(AI)模型的发展——从简单、专门化、浅层模型发展到深度、多模态、通用模型的过程

a部分:在2000年代初,医学影像中AI技术的进展开始广泛使用监督学习,利用手工设计的特征的机器学习模型。这些模型依赖于领域专家从图像中手动提取相关特征,然后用于在标记数据集上训练模型。到了2012年左右,监督深度学习出现,AI模型,特别是卷积神经网络(CNNs),在大型标记数据集上进行训练,以直接从原始图像数据自动学习分层特征。这种方法显著提高了AI模型在医学影像任务中的性能和泛化能力。在2020年代初,许多研究小组开始使用新兴的自监督学习方法,这些方法使模型能够通过预测数据本身的属性,而不是依赖外部标签,从未标记的数据中学习有意义的模式。
b部分:在AI应用于癌症研究的初期阶段,方法是依赖于医学图像中精心选择的模式作为分析的基础。随着时间的推移,手动特征选择的需求被消除,模型能够直接从数据中学习基本特征。目前,重点是通过多模态模型整合不同的数据源,这些模型结合了不同数据模态的信息,如放射图像、病理幻灯片、基因组数据和临床记录。设想的未来涉及基础模型,这些是大规模的自监督模型,预先在多种未标记的数据集上进行预训练,涵盖多种模态。这些模型可以针对各种下游任务进行微调,只需最少的任务特定训练数据。AI在肿瘤学应用的最终目标可能是一个通用的通用模型,这是一个多用途工具,能够分析、解释并与患者和医疗专业人员互动。这个通用模型将整合来自多个来源的数据,支持诊断和治疗建议,并以人类可解释的方式解释其决策。
癌症研究中基于文本的假设性人工智能(AI)工作流程
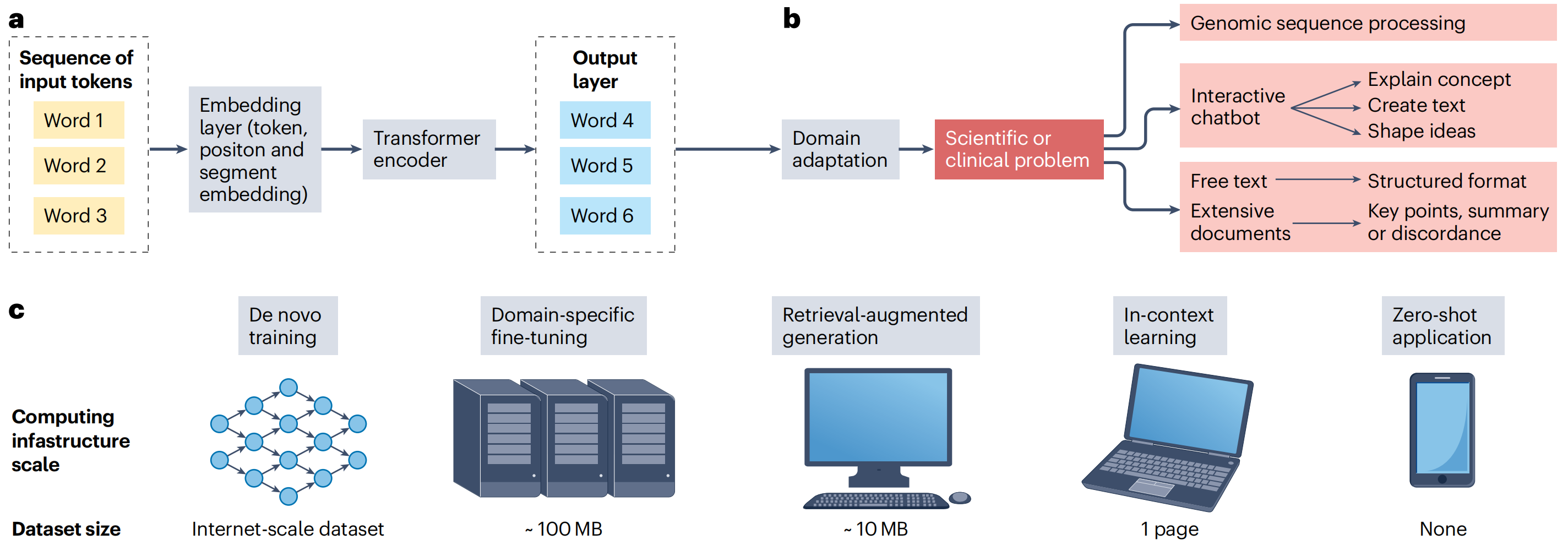
a部分:描述了自然语言处理(NLP)模型的演变,从循环神经网络(RNN)架构转变为变换器(transformer)架构。在变换器模型中,输入的词元(例如单词)首先通过NLP模型的一个关键部分——嵌入层(embedding layer)转换为向量。这里,单词或短语被转换为数值表示,使模型能够处理语言。这些向量连同位置和段落嵌入(它们增加了关于词元顺序和关系的上下文信息)一起被传递到“变换器编码器”(transformer encoders)。变换器编码器是模型的高级组件,通过考虑每个单词相对于其他单词的上下文来处理输入,使模型能够理解复杂的语言模式。变换器模型的输出层根据处理过的输入生成最终的预测或表示,这些可以用于各种下游任务,如文本分类、命名实体识别或文本生成。
b部分:展示了训练有素的NLP模型可以应用于癌症研究的多种任务。例如,它们可以将非结构化的临床笔记或研究论文转换为结构化格式,使信息更易于访问和分析。这可以通过在大量标注文本数据上训练模型来实现,其中所需的结构化格式被提供为标签。模型学习识别关键信息并将其映射到结构化格式中的适当字段。NLP模型还可以总结长篇文档,如研究文章或临床试验报告,突出关键点和发现。
c部分:讨论了这些模型的训练和微调取决于数据集大小和计算能力。从头开始训练大型语言模型(LLMs)需要包含数十亿个单词的互联网规模数据集和庞大的计算基础设施。然而,一旦这些大型模型预训练完成,它们就可以使用更小的领域特定数据集进行特定任务的微调,通常在几百到几千个示例的范围内。这个微调过程计算要求较低,可以在较小规模的基础设施上执行。对于某些任务,LLMs可以以“零样本”(zero-shot)的方式使用,它们执行任务时不需要任何额外的训练,只依赖于它们已有的知识。这种零样本能力允许LLMs应用于广泛的任务,甚至在资源受限的设备上,如手机。上下文学习(in-context learning)和检索增强生成(RAG)是替代方法,模型作为输入提示的一部分提供一些期望任务的示例,或通过提供额外的文档,分别使模型能够适应任务而无需明确的微调。这些不同的方法使语言理解和生成能力更易于访问和高效。
三、跨物种心脏移植:当猪心在人体内跳动
文章概述
这篇文章是发表在《Nature Medicine》上的一项研究,标题为“Integrative multi-omics profiling in human decedents receiving pig heart xenografts”。研究的主要目的是通过多组学分析,来更好地理解人类受体在异种心脏移植(即从基因编辑过的猪到人类)后的分子景观。
研究团队对两位已故人类受体(D1和D2)进行了心脏异种移植,并在移植后的72小时内,每6小时收集一次血液样本,进行了大规模的转录组学、脂质组学、蛋白质组学和代谢组学分析。此外,还对受体的组织样本进行了组织学和转录组学分析。
研究的设计和样本收集时间线

上图详细说明了两位已故受体(decedents,D1和D2)在心脏异种移植后样本的采集计划以及随后的分析流程。
- 样本采集:
- 在移植后66小时,两位受体的左心室和右心室组织样本被采集用于单核RNA测序(snRNA-seq)和空间转录组分析。
- 外周血样本则在移植后的每个6小时间隔收集一次,用于进行包括代谢组学、蛋白质组学、脂质组学、细胞因子分析、批量RNA测序(bulk RNA-seq)、单细胞RNA测序(scRNA-seq)和流式细胞术在内的全面分析。
- 特定样本分析:
- 对于D2受体,66小时时的组织样本通过scRNA-seq和bulk RNA-seq进行了分析。
- 对于D1受体,在64小时、65小时和65.5小时专门收集了血液样本。
- 血液检测时间线:
- 图表的底部展示了移植后血液检测的执行时间线。
- 灰色点表示两个受体都进行了采样的时间点。
- 紫色或绿色点表示仅对D1或D2受体进行采样的时间点。
- 临床常规测量:
- 通过血液测量了包括国际标准化比率(INR)、丙氨酸转氨酶(ALT)和天门冬氨酸转氨酶(AST),以及动脉血气、全面代谢组、完全血细胞计数、乳酸脱氢酶和肌钙蛋白水平等常规临床指标。
研究发现,在D1受体中观察到了显著的早期免疫反应,特别是在外周血单核细胞和异种移植物组织中,与T细胞和自然杀伤细胞活性相关。纵向分析显示,存在缺血再灌注损伤,这种损伤由于T细胞免疫抑制不足而加剧,与之前在猪到非人灵长类动物研究中发现的围手术期心脏异种移植功能障碍一致。此外,在移植后42小时,D1受体发生了细胞代谢和肝脏损伤途径的显著变化,与全身器官功能障碍相关。
相比之下,D2受体的RNA、蛋白质、脂质和代谢组学特征变化较小。这些多组学分析揭示了两位人类受体对心脏异种移植的不同反应,并为早期分子和免疫反应提供了新的见解。这些发现可能有助于开发针对性的治疗方法,以限制缺血再灌注损伤相关表型,并改善治疗结果。
文章还讨论了异种移植在解决人类同种移植中供体器官严重短缺问题方面的潜力。通过使用最近死亡的脑死亡人类供体和关键异种抗原(包括α-1,3-半乳糖转移酶)的基因敲除模型,异种移植研究已经从临床前灵长类动物实验过渡到人类。
这项研究是首次在心脏异种移植受体中进行如此密集的时间序列多组学分析,为理解异种移植后的分子动态提供了新的视角,并强调了在所有未来的异种移植案例中,尤其是在早期时间点,使用详细的综合分子监测的重要性。
重点关注
空间转录组学分析揭示的血管重塑和缺血情况
空间转录组学切片,通过斑点对应于内皮信号进行了映射。基于表达信息,对空间转录组数据进行了聚类分析。在D1的左心室(左侧)和D2的右心室(右侧)中,标记出了表达内皮和血管标志物的空间簇(簇5、7、8和9)。每个样本有两个技术重复(A和B)。D1-LV-A代表D1的左心室,重复A;D2-RV-A代表D2的右心室,重复A。比例尺,1毫米。

D1的猪心脏异种移植内膜心肌活检组织的H&E染色,显示内皮细胞“抬起”(白色箭头),存在少量炎症细胞(黑色和蓝色箭头)。比例尺,1毫米。
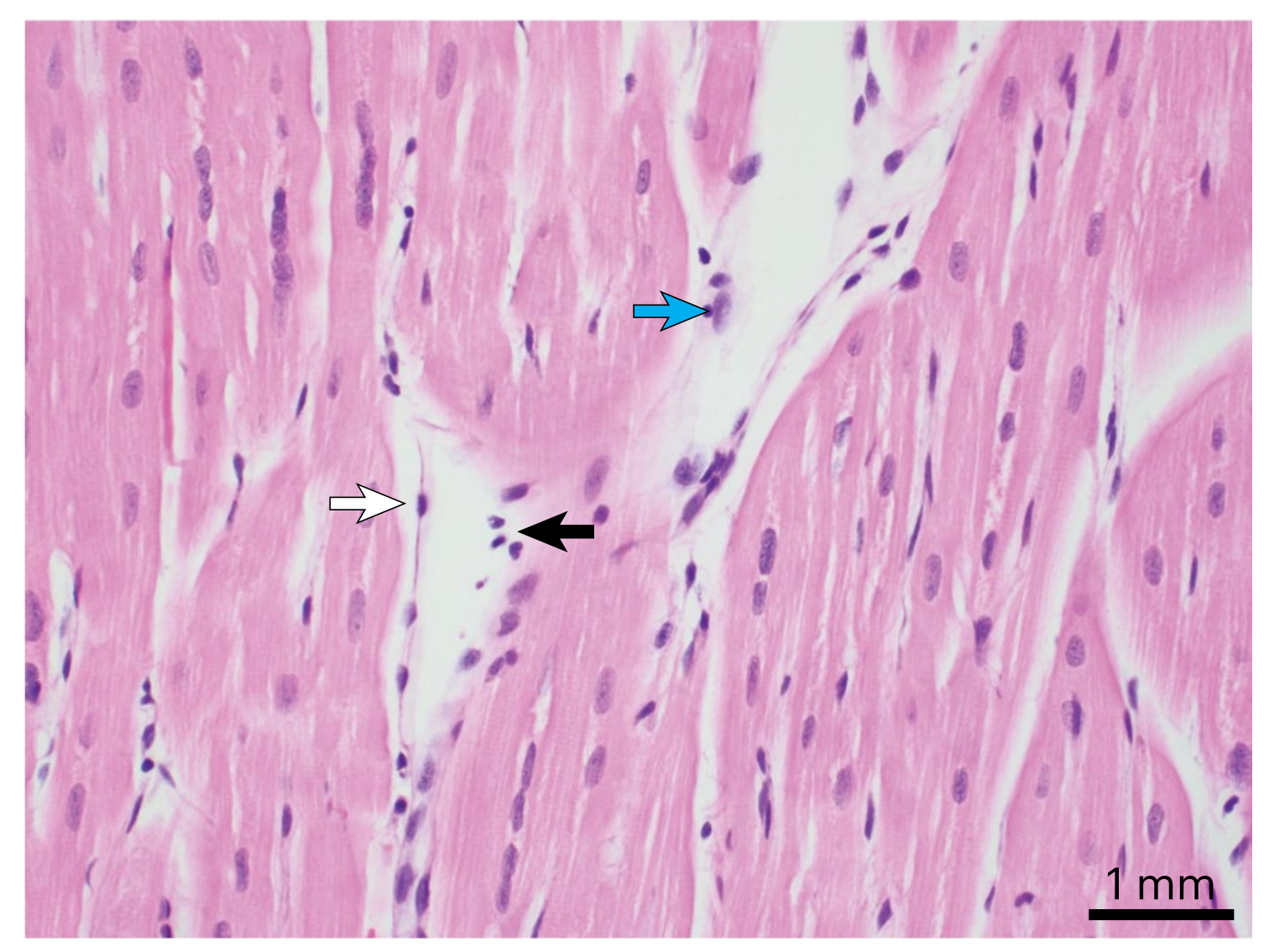
来自D1异种移植样本的电子显微镜图像,显示内皮细胞肿胀(白色箭头)和肌膜破裂(红色箭头)。比例尺,5微米。

四、Nature Medicine·DEPLOY模型:从病理学图像直接预测DNA甲基化,破解CNS肿瘤诊断难题!

文献概述
这篇文章是发表在《Nature Medicine》上的研究,标题为“Prediction of DNA methylation-based tumor types from histopathology in central nervous system tumors with deep learning”。研究团队开发了一个名为DEPLOY(Deep lEarning from histoPathoLOgy and methYlation)的深度学习模型,用于从组织病理学图像中分类中枢神经系统(CNS)肿瘤到十个主要类别。
研究背景:
- CNS肿瘤类型多样,精确诊断对治疗至关重要。
- DNA
甲基化分析可以提高诊断准确性,但过程耗时且不普遍可用。
DEPLOY模型:
-
包含三个独立组件:
- 直接从幻灯片图像分类肿瘤的“直接模型”;
- 首先生成DNA甲基化beta值预测,然后用于肿瘤分类的“间接模型”;
- 直接从常规可用的患者人口统计数据分类肿瘤类型的“人口统计模型”。
-
通过
内部数据集训练(1796名患者),在三个独立的外部测试数据集上验证(共2156名患者),DEPLOY模型在高置信度预测样本上达到了95%的总体准确率和91%的平衡准确率。
研究结果:
- DEPLOY能准确从组织病理学图像预测beta值。
- 在多中心验证中表现出色,证明了其作为病理学家辅助诊断工具的潜力,尤其是在资源有限的地区。
研究意义:
- 提供了一种快速、准确的
CNS肿瘤分类方法。 - 为未来开发高分辨率基于H&E图像的肿瘤分类器奠定了计算基础,可能扩展到其他癌症类型的分类。
研究限制:
- 需要匹配的组织病理学图像和甲基化数据来开发新肿瘤类型的预测器。
- 当前模型训练主要针对
十个主要肿瘤类型,但实际应用中可能需要进一步扩展到更多类型。
未来展望:
- 随着数据集的扩大,DEPLOY的性能有望进一步提升。
- 该模型可能成为全球病理学家的辅助工具,尤其是在资源不足的地区提供帮助。
整体而言,这项研究展示了深度学习在提高CNS肿瘤诊断准确性方面的潜力,尤其是在传统方法受限的情况下。
重点关注
DEPLOY 模型的数据集和计算工作流程

a. 患者队列:
- DEPLOY 使用内部国家癌症研究所(NCI)的数据集进行训练和交叉验证,该数据集包含 1796 名患者的匹配幻灯片和甲基化档案。
- 为了外部验证,研究者利用了来自数字脑肿瘤图谱(DBTA)的 1522 名患者、儿童脑肿瘤网络(CBTN)的 348 名患者以及 NCI 前瞻性队列的 286 名患者的幻灯片。
b. DEPLOY 框架的关键组件:
- 最初,整个幻灯片图像(WSIs)被划分为瓦片(tiles)。
- 这些瓦片使用预训练的 ResNet50 模型进行特征提取。
- 接着,使用自动编码器将 2048 个 ResNet 特征压缩到 512 维的低维表示。
- 为了构建 DNA 甲基化模型,基于自动编码器特征使用多层感知器(MLP)回归。
- DEPLOY 结合了三种模型进行肿瘤分类:
- 直接模型(橙色方块):使用自动编码器压缩的特征,通过 MLP 分类器直接预测肿瘤类型。
- 间接模型(绿色方块):使用四种经典的机器学习算法根据预测的 DNA 甲基化水平对肿瘤进行分类。
- 人口统计模型(浅蓝色):与间接模型使用相同的四种分类器,结合年龄、性别和肿瘤位置信息进行肿瘤类型分类。
- 综合模型:通过计算这三种方法的预测分数的平均值来综合预测,无需额外的训练或微调。
c. 值得注意的是:
- 虽然类别标签是在全幻灯片级别上,但 DEPLOY 的直接和间接模型能够提供瓦片级别分辨率的预测。
- 这有时可以预测单个 WSI 中以空间有序方式描绘的不同肿瘤类型的共存(见图 5)。
图中还列出了 CNS 肿瘤的十种类型,包括:
- A-IDH:IDH 突变型星形细胞瘤
- CP:脉络丛
- EPEN:室管膜瘤
- GBM:胶质母细胞瘤
- MB:髓母细胞瘤
- MEN:脑膜瘤
- MPE:粘液性室管膜瘤
- O-IDH:IDH 突变型少突胶质细胞瘤
- PA:毛细胞星形细胞瘤
- SE:亚室管膜瘤
这个流程图说明了 DEPLOY 模型如何通过深度学习从组织病理学图像中预测 DNA 甲基化状态,并据此对肿瘤类型进行分类,同时也展示了如何整合不同的数据和模型来提高分类的准确性。
在一组诊断具有挑战性的病例中,DEPLOY 模型提出的诊断变化

这些肿瘤来自 NCI 队列,其中 DEPLOY 的预测与最初的病理学家诊断不同,但与甲基化类别一致。
a. 三个临床类别中肿瘤的比例和数量:
- 这部分通过条形图表示,不同颜色的条形代表不同的临床影响级别:
- 简单诊断变化(可能涉及诊断术语的轻微变化,临床影响有限)
- 确定性诊断的建立(从一般描述性标签变为明确的肿瘤类别,具有临床相关性)
- 具有临床影响的诊断变化(涉及肿瘤分级和/或患者管理计划的潜在变化)
- 条形图显示了每种变化类别中的肿瘤数量和比例。
b. Sankey 图显示了 DEPLOY 建议的诊断变化(并通过甲基化分类验证):
- Sankey 图是一种流程图,显示了从原始诊断到 DEPLOY 预测的诊断变化流程。
- 图中的颜色与 a 部分中的颜色相对应,以示区分不同类型的诊断变化。
- 包括了各种肿瘤类型,如脉络丛(CP)、脑膜瘤(MEN)、粘液性室管膜瘤(MPE)、亚室管膜瘤(SE)、多形性黄色星形细胞瘤(PXA)、未另行指明(NOS)、具有毛发样特征的高级别星形细胞瘤(HGAP)、神经节胶质瘤(GG)和松果体母细胞瘤(PB)。
c. 树图对比了 DEPLOY 的分类与原始病理学家的诊断:
- 树图显示了 DEPLOY 预测与甲基化类别匹配的案例数量和比例。
- 顶部的树图(teal 色)显示了 DEPLOY 的 top one 预测与甲基化类别匹配的案例。
- 中间的树图(灰色)显示了 DEPLOY 的 top two 预测与甲基化类别匹配的案例。
- 底部的树图(粉色)显示了 DEPLOY 的 top one 和 top two 预测均与甲基化类别不一致的案例。
- 上部树图展示了所有 309 个 DEPLOY 预测类别与最初病理学家诊断不同的案例。
- 下部树图专注于诊断具有挑战性的胶质瘤的子集,包括胶质母细胞瘤、毛细胞星形细胞瘤(PAs)和 IDH 突变胶质瘤(详见方法部分的 “诊断具有挑战性的胶质瘤分类” 章节)。
总体而言,上图强调了 DEPLOY 模型在病理诊断中的潜在作用,特别是在那些最初诊断难以确定的复杂病例中。通过与甲基化类别的一致性验证,DEPLOY 显示出可能改善诊断准确性和临床管理的能力。
以一个双基因型的少突胶质细胞瘤(oligoastrocytoma)的分析为例,展示了DEPLOY 模型如何提供空间甲基化预测&对肿瘤异质性进行评估
a. H&E染色肿瘤观察:
- 展示了一个 H&E 染色的肿瘤图像,其中有少突胶质瘤和星形细胞瘤的组织学区域。
- 图中用方框1和方框2标记了两个特定感兴趣的区域。
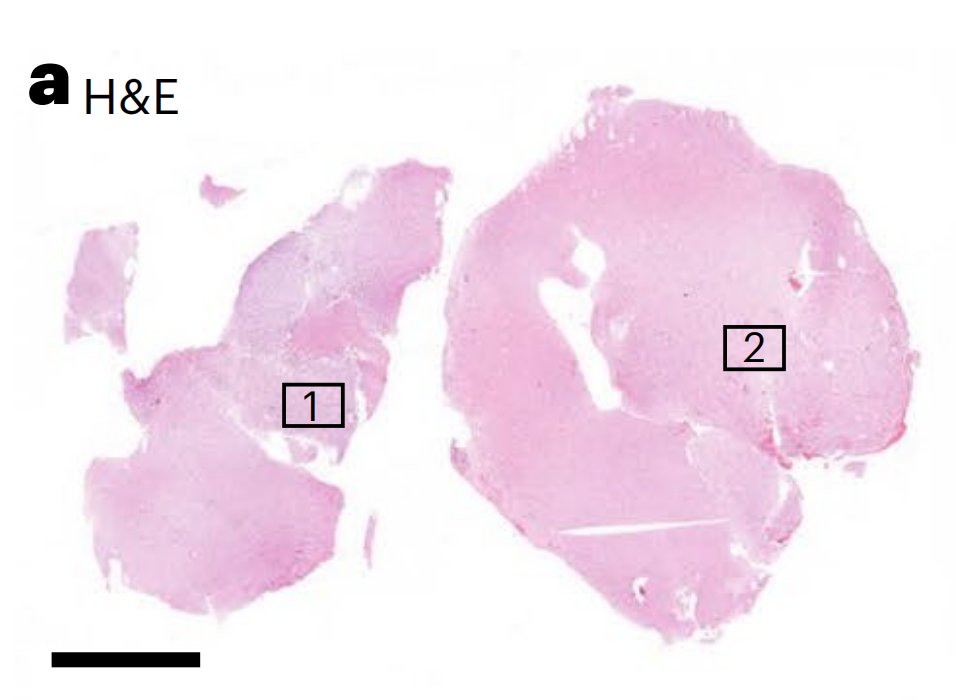
b. DEPLOY 肿瘤类型预测(瓦片级别):
- 瓦片级别的 DEPLOY 预测显示,蓝色代表预测为少突胶质瘤,红色代表预测为星形细胞瘤。
- 这表明 DEPLOY 能够在单个幻灯片的不同区域中区分不同的肿瘤类型。

c. IDH1-R132H的免疫组化:
- 展示了针对 IDH1-R132H(胶质瘤中的 IDH 突变)的免疫组化图像。
- 这种突变通常与星形细胞瘤相关。
d. ATRX的免疫组化:
- 展示了针对 ATRX 的免疫组化图像。
- ATRX 通常在所有细胞中存在,但在 IDH 突变的星形细胞瘤中丢失,在少突胶质瘤中保留。

五、治疗抵抗性肿瘤的克星:发现垂体肿瘤的新型生物标志物

文献概述
这篇文章是关于垂体神经内分泌肿瘤(PitNETs)的研究,特别是那些表现出侵略性、难以治疗的肿瘤。研究团队由Andrew L. Lin、Vasilisa A. Rudneva、Allison L. Richards等多位作者组成,他们对两组PitNETs患者进行了基因组测序:一组是手术前同意参加测序的前瞻性患者(66名),另一组是回顾性患者(26名),包括那些表现出侵略性/高风险PitNETs的患者。
研究发现,与良性肿瘤相比,侵略性、难以治疗的PitNETs具有更高的突变负担和杂合性丧失(LOH)的比例。特别是在类固醇细胞系中,12条特定染色体上反复出现的染色体LOH模式与治疗抵抗性相关。在整个队列中,TP53突变的肿瘤中识别出更高的LOH比例。通过机器学习方法,研究确定了LOH是预测侵略性、难以治疗行为的最具有预测性的变量,其准确性为0.88(95% CI: 0.70–0.96),超过了最常见的基因水平改变TP53。
文章强调,侵略性、难以治疗的PitNETs的特征是显著的非整倍性,这是由广泛的染色体LOH引起的,特别是在类固醇细胞肿瘤中。这种LOH可以以高精度预测治疗抵抗性,并且代表了这一定义不清的PitNET类别的一个新的生物标志物。
文章还详细介绍了研究的材料和方法,包括患者的选择、免疫组化、基因组测序和分析、荧光原位杂交(FISH)实验以及机器学习的使用。此外,还讨论了统计分析和研究结果,包括患者的人口统计数据、基因水平的反复体细胞改变、治疗抵抗性PitNETs中的反复基因组广泛LOH以及未来治疗抵抗行为的基因组标记的随机森林建模。
最后,文章讨论了研究的意义,指出目前缺乏预测未来侵略性、难以治疗行为的生物标志物,并强调了LOH作为一个新的生物标志物的重要性,它可能对治疗这种罕见恶性肿瘤具有重要意义。
重点关注
侵袭性、治疗难治性(aggressive, treatment-refractory)和良性(benign)垂体神经内分泌肿瘤(PitNETs)的基因组变异和临床特征

(a) 回顾性和前瞻性组的临床行为关系图
这部分是一个示意图,展示了回顾性组和前瞻性组之间的关系以及它们的临床行为。回顾性组包括了已经接受过治疗并表现出特定临床行为的患者,而前瞻性组则是在未来的研究中将被跟踪观察的患者群体。
(b) 肿瘤谱系总结和转移疾病状态
这部分提供了治疗难治性和良性肿瘤亚组的肿瘤谱系(即肿瘤细胞的起源和类型)总结,以及在数据截止时(治疗难治性亚组)的转移疾病状态。这可以帮助理解不同肿瘤类型和它们转移的倾向性。
© 肿瘤驱动基因的Oncoprint总结
Oncoprint是一种可视化方法,用于展示肿瘤中反复改变的驱动基因。在这个图中,左侧(n=23)代表治疗难治性PitNETs,右侧(n=69)代表良性PitNETs。如果患者有多个肿瘤样本,将展示所有样本中变异的联合集。图中的上方列出了患者的人口统计学和临床病理特征,下方列出了常见的基因变异(右侧以百分比显示每个临床类别的频率)。如果进行了全外显子重捕获测序,还会报告USP8基因的状态。
六、AI解码生命:如何用一张图片预测卵巢癌治疗效果?

全文概述
这篇文章是关于一种名为Pathologic Risk Classifier for High-Grade Serous Ovarian Cancer (PathoRiCH)的新型深度学习分类器的研究。该分类器是基于组织病理学图像开发的,用于预测高级别浆液性卵巢癌(HGSOC)患者对铂基化疗的反应。目前,没有可用的生物标志物能够迅速预测对铂基治疗的反应,因此这项研究旨在填补这一空白。
图1提供了研究中使用的多实例学习(Multiple Instance Learning, MIL)模型,使用了不同放大倍数(5倍和20倍)的组织切片图像(Whole-Slide Images, WSIs)的图像块(patches)。

以下是该图内容的详细解释:
-
图像块提取:首先从WSIs中提取了不同放大倍数的图像块。5倍和20倍代表了不同的细节级别,5倍提供组织结构级别的视图,而20倍提供细胞级别的细节。
-
自动化癌症分割:接着,使用自动化癌症分割模型处理这些图像块,以排除不包含癌细胞的图像块。这个分割模型预先使用侵袭性
乳腺导管癌的图像进行了训练。 -
对比自监督学习算法:通过分割的癌症区域图像块被输入到一个
对比自监督学习算法中(图中蓝色箭头路径)。这种算法可以帮助模型学习图像的特征表示,而不需要外部的标签信息。 -
全组织学习:或者,所有的图像块,包括那些不包含癌细胞的图像块,可以直接输入到自监督学习算法中,以包含WSIs中的所有组织(图中红色箭头路径)。
-
多实例学习方法:对于单一尺度和多尺度放大设置(即5倍、20倍和两者结合),分别使用了不同的MIL方法。这样,生成了六个不同的MIL模型。
-
多尺度MIL:在多尺度MIL模型中,通过将不同尺度的WSIs的嵌入(embeddings)连接起来形成特征金字塔(feature pyramids),以此来训练MIL聚合器。这种方法可以整合不同尺度的信息,以提高模型的性能。
总的来说,图1展示了一个综合的深度学习框架,它结合了图像分割、自监督学习和多实例学习技术,以预测高级别浆液性卵巢癌对铂基化疗的反应。通过这种方法,研究者能够开发出能够从组织病理图像中自动学习和预测临床结果的强大模型。
研究人员在内部队列(n=394)上训练了PathoRiCH,并在两个独立的外部队列(n=284和n=136)上进行了验证。
表1提供了在研究中包括的三个不同队列(SEV、TCGA和SMC)的高级别浆液性卵巢癌(HGSOC)患者的临床病理特征的总结。
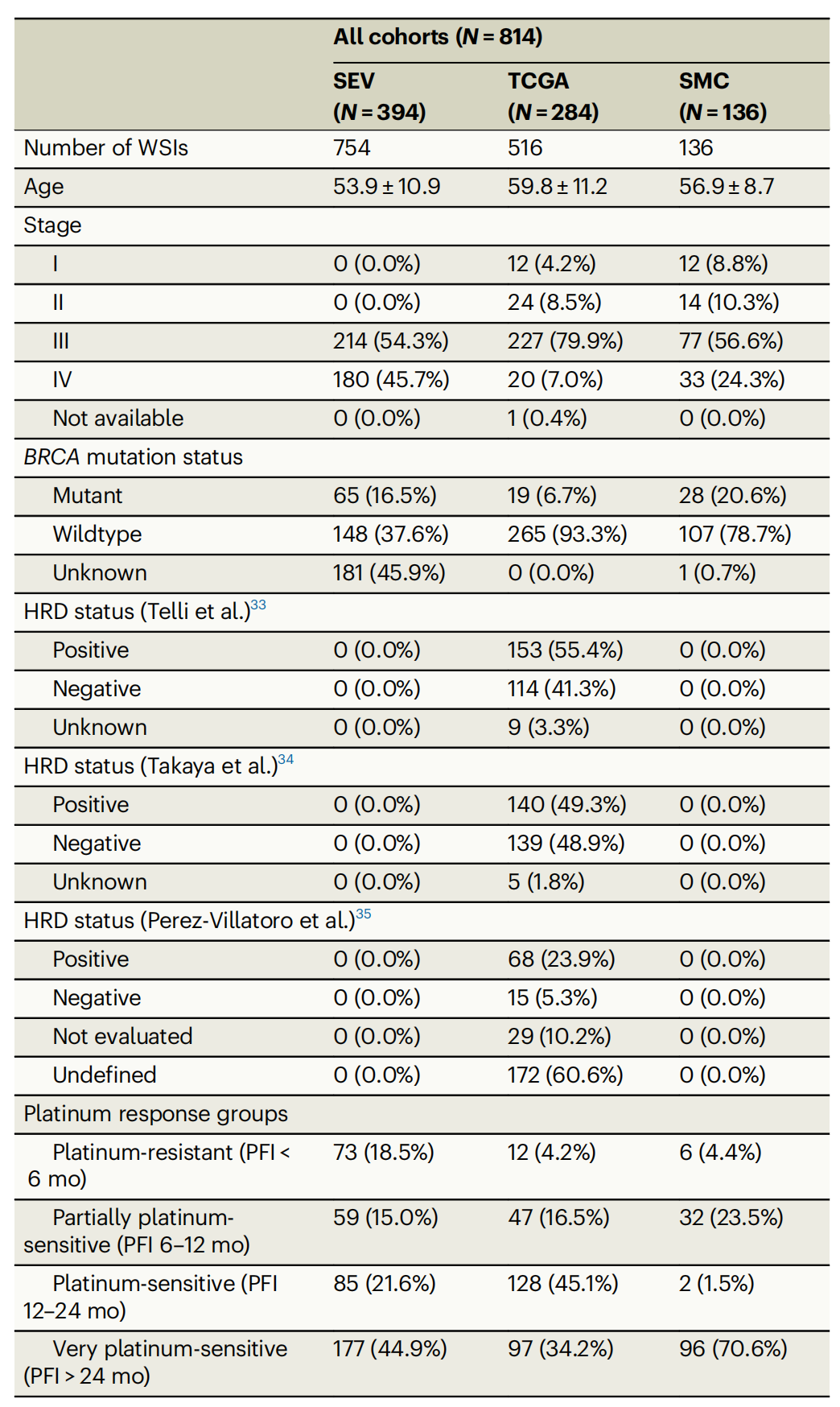
这些特征包括:
- Number of WSIs: 每个队列中全切片图像(Whole Slide Images,WSIs)的数量。
- Age: 患者的平均年龄以及标准差。
- Stage: 患者的癌症分期,从I到IV,分期越高表示癌症越晚期。
- BRCA mutation status: 患者的BRCA基因突变状态,包括突变型(Mutant)、野生型(Wildtype)和未知(Unknown)。
- HRD status: 同源重组缺陷(Homologous Recombination Deficiency,HRD)状态,根据Telli等人、Takaya等人和Perez-Villatoro等人的研究分别列出,包括阳性、阴性和未知。
- Platinum response groups: 根据铂无进展间期(Platinum-Free Interval,PFI)对患者进行分类,分为铂类抗药性(Platinum-resistant,PFI ≤ 6个月)、部分铂类敏感性(Partially platinum-sensitive,PFI 6–12个月)、铂类敏感性(Platinum-sensitive,PFI 12–24个月)和非常铂类敏感性(Very platinum-sensitive,PFI > 24个月)。
表格中列出的具体数字表示每个分类下的患者数量,以及对于某些分类(如BRCA突变状态和HRD状态)的百分比。这个表格是理解研究人群特征的基础,并且对于分析和比较不同队列间的差异以及它们对研究结果的潜在影响至关重要。
结果显示,PathoRiCH预测的有利反应组和不良反应组在所有三个队列中的铂无进展间期(platinum-free intervals, PFI)有显著差异。
将PathoRiCH与分子生物标志物结合使用,可以为患者的风险分层提供更强大的工具。通过可视化和转录组分析解释了PathoRiCH的决策过程,增强了模型决策的可靠性。PathoRiCH显示出比当前分子生物标志物更好的预测性能,为开发创新工具以转变HGSOC的当前诊断流程提供了坚实的基础。
研究还涉及了多种多实例学习(MIL)模型,这些模型仅使用组织病理学图像来预测女性HGSOC患者对铂基治疗的反应。通过这种方法,研究人员能够开发出一个稳健的MIL模型,即PathoRiCH。PathoRiCH在训练的HGSOC队列中表现出显著的预测性能。此外,将PathoRiCH与当前的分子生物标志物结合,为HGSOC患者的更精确风险分层方法提供了可能。
文章还讨论了PathoRiCH的独立预后因素,并通过可视化分析和转录组分析进一步验证了模型的可靠性。研究结果表明,PathoRiCH可以作为HGSOC患者个体化治疗的临床相关信息提供者。
最后,文章讨论了PathoRiCH的局限性和未来改进的方向,包括需要在更多的中心进行验证,以及需要进一步解释模型以确保其稳健性并引入临床实践。研究还提到了需要平衡训练数据集中对反应不良的病例,以改善模型训练。此外,研究还提到了对BRCA突变和HRD状态预测的模型性能进行了评估,并指出需要大型BRCA和HRD专注的队列来改进模型。
重点关注
图3展示了在TCGA(The Cancer Genome Atlas)外部验证队列中,结合PathoRiCH、BRCA和HRD(同源重组缺陷)结果对患者进行分类的Kaplan-Meier生存分析和真实的铂无进展间期(PFI)组的分布情况。

a部分:
- Kaplan-Meier生存曲线: 这部分展示了根据PathoRiCH、BRCA和HRD的综合结果对患者进行分类后的生存曲线。生存曲线通常用来表示生存时间的分布情况。
- 显著性差异: 结合PathoRiCH、BRCA和HRD的结果在PFI(铂无进展间期)和OS(总生存期)上显示出显著的差异,p值分别为1.07E-05和3.30E-16,这表明这种分类方法能够有效地区分患者的生存结果。
- 不同组的生存情况:
- 有利的BRCA/HRD阳性组(favorable–BRCA/HRD-positive group)显示出最有利的PFI。
- 不利的BRCA/HRD阳性组(poor–BRCA/HRD-positive)和不利的BRCA/HRD阴性组(poor–BRCA/HRD-negative)显示出最差的PFI。

b部分:
- PFI组的分布: 这部分展示了四个PFI组的分布情况,包括
铂类药物抗性(PFI ≤ 6个月)、部分铂类药物敏感性(6–12个月)、铂类药物敏感性(12–24个月)和非常铂类药物敏感性(>24个月)。 - 柱状图: 柱状图用不同颜色(
蓝色代表有利,红色代表不利)表示每个结果组的预测百分比,每个柱状图内部的数值显示了每个类别的案例数量。 - 显著性检验: 结合PathoRiCH、BRCA和HRD的分类显示出四个PFI组的分布有显著性差异,p值为0.001。
整体而言,图3强调了PathoRiCH结合分子生物标志物(BRCA和HRD状态)在预测HGSOC患者对铂基化疗反应和生存结果方面的强大能力。通过这种综合分类方法,研究人员能够更精确地对患者进行风险分层,并可能指导更个性化的治疗决策。
在TCGA和SMC两个外部验证队列中进行的多变量Cox回归分析的结果

分析使用以下六个变量:
- 年龄(Age)
- 国际妇产科联盟分期(FIGO stage),用于评估癌症的严重程度和扩散情况
- BRCA基因突变状态(BRCA mutation status)
- 同源重组缺陷(HRD)状态
- 结合BRCA和HRD状态
- PathoRiCH预测结果
数据呈现:
- 结果以
误差条(error bar)表示,代表95%置信区间(confidence interval)。置信区间越窄,表示估计越精确。 - Cox回归分析的结果通常包括每个变量的风险比(hazard ratio)和对应的p值。
a部分(TCGA队列):
- 在TCGA队列中,PathoRiCH预测结果是最强的独立预后因素,具有显著的统计学意义(p = 6.57E-05),表明PathoRiCH预测对患者生存时间的预测非常有效。
- FIGO分期也是一个显著的预后因素(p = 0.005),意味着癌症的严重程度和扩散情况对患者的生存有重要影响。
- BRCA状态(p = 0.32)在这个队列中也显示出一定的影响,但并不像PathoRiCH和FIGO分期那样显著。
b部分(SMC队列):
- 在SMC队列中,FIGO分期(p = 0.004)和PathoRiCH(p = 0.039)显著地作为独立的预后因素。尽管PathoRiCH的p值不如TCGA队列中的显著,但仍然表明它是一个重要的预后因素。
- 值得注意的是,PathoRiCH在SMC队列中的p值较高(p = 0.039),这可能意味着在该队列中PathoRiCH预测的生存影响不如在TCGA队列中那么显著,或者需要更大的样本量来确认其影响。
PathoRiCH预测出的有利和不利两组患者的注意力图谱分析

注意力图谱是一种可视化技术,它可以帮助我们理解深度学习模型在做出预测时关注的图像区域。
左侧部分:
- Kaplan-Meier生存分析: 图的左侧展示了根据PathoRiCH预测结果进行的双边Kaplan-Meier生存曲线分析。这种分析通常用来展示不同组别患者的生存率随时间的变化情况。
- 生存曲线: 曲线显示了有利组和不利组患者的生存差异,从而验证PathoRiCH预测的有效性。
注意力图谱:
- 单独与组合注意力图: 为有利和不利预测分别创建了单独的注意力图,然后将这些图组合起来,为每个患者生成了一个综合的预测注意力图。这可以帮助识别模型在做出特定预测时所侧重的图像区域。
- 比例尺: 注意力图的比例尺为2毫米(scale bar = 2 mm),这提供了图像中显示区域的实际大小参考。
- 代表性案例: 图中展示了来自有利组和不利组的两个代表性患者的案例。
- 高得分区域: 对应的注意力图和高得分区域(high-score patches)并排显示,比例尺为50微米(scale bar = 50 µm)。高得分区域是模型特别关注并用于做出预测的图像部分。
注意力图的作用:
- 注意力图谱分析可以揭示模型预测背后的模式和特征,比如在病理图像中,模型是否更多地关注了肿瘤的某些特定形态学特征。
- 通过观察高得分区域,研究人员可以了解模型认为与治疗反应有利或不利相关的病理学特征。
总的来说,图5提供了一个直观的方法来理解PathoRiCH模型是如何通过分析病理图像并集中关注某些区域来预测HGSOC患者的治疗反应的。这种分析有助于提高模型的透明度和可信度,同时也可能为病理学家提供有价值的洞察,帮助他们理解影响患者治疗反应的潜在生物标志物。
















 6302
6302











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









