







小罗碎碎念
本期推文主题:人工智能在肿瘤生物标志物领域的最新研究进展
今天这期推文虽然主要介绍的是与生物标志物相关的内容,但是研究病理AI的老师也不要错过,因为有两篇内容直接与病理相关——第一篇和第五篇。
第一篇介绍的是病理AI领域目前最新最大的基础模型,发表于7-22。截止今日发文,我已经在推文中推荐过三篇病理AI的基础模型,明日推文会先做一个汇总,后续推文再介绍如何利用基础模型缩短自己的训练过程;第五篇是病理AI的一个多模态模型,更精确的说,是双模态模型,因为它只结合了临床信息和WSI,研究的是生物标志物在高风险前列腺癌预后评估中的作用。
最后提一嘴第六篇文献,这篇文章用的是全基因组测序的数据,我看到以后的第一反应——单细胞数据是不是也可以按照这个套路来弄?所以我也在这一部分简要介绍了单细胞测序与全基因组测序的区别,欢迎各位专业领域的老师/同学探索这一方案的可行性。

一、泛癌种检测与生物标志物预测:Virchow基础模型的性能与潜力

一作&通讯
| 作者类型 | 作者姓名 | 单位名称(英文) | 单位名称(中文) |
|---|---|---|---|
| 第一作者 | Eugene Vorontsov | Paige, New York, NY, US | 佩奇公司,纽约,美国 |
| 第一作者 | Alican Bozkurt | Microsoft Research, Cambridge, MA, US | 微软研究院,剑桥,马萨诸塞州,美国 |
| 通讯作者 | Siqi Liu | Paige, New York, NY, US | 佩奇公司,纽约,美国 |
| 通讯作者 | Thomas J. Fuchs | Memorial Sloan Kettering Cancer Center, New York, NY, US | 纪念斯隆凯特琳癌症中心,纽约,美国 |
文献概述
这篇文章介绍了Virchow,一个用于临床级计算病理学和罕见癌症检测的新型大型基础模型,它通过自监督学习在大量病理图像数据上训练,在多种癌症检测和生物标志物预测任务中展现出高性能。
Virchow是目前最大的计算病理学基础模型,它不仅能够预测生物标志物和识别细胞,还能实现泛癌种检测,包括九种常见和七种罕见的癌症类型。研究显示,即使在训练数据较少的情况下,基于Virchow构建的泛癌种检测器也能够达到与特定组织临床级模型相似的性能,并且在某些罕见癌症变体上表现更优。
Virchow模型通过自监督学习算法训练,能够生成数据表示(嵌入),这些嵌入能够很好地泛化到多种预测任务。与传统的计算病理学方法相比,Virchow能够捕捉到更广泛的组织形态学变化和实验室准备的变化,这对于临床实践中的泛化至关重要。此外,Virchow模型在处理罕见肿瘤类型或不常见的诊断任务(如预测特定的基因组变化、临床结果和治疗反应)时,显示出了其独特的价值。
研究还展示了Virchow在生物标志物预测方面的应用,这可以减少对额外测试的需要,加快为患者提供关键数据的速度。Virchow模型在多个层面上展示了其在计算病理学新领域的强大潜力,包括在罕见癌症检测和生物标志物预测方面的性能。
文章还讨论了Virchow模型的潜在临床影响,包括在临床实践中减少诊断周转时间、为不常见癌症开发临床级产品、以及使用常规H&E染色的WSI进行生物标志物预测等。
此外,文章也指出了Virchow模型的局限性,包括训练数据集来源单一、模型和数据规模的饱和点尚未明确等,并对未来的研究方向提出了展望。
重点关注
图1提供了Virchow模型研究的全面概述,包括训练数据集、训练算法和应用。
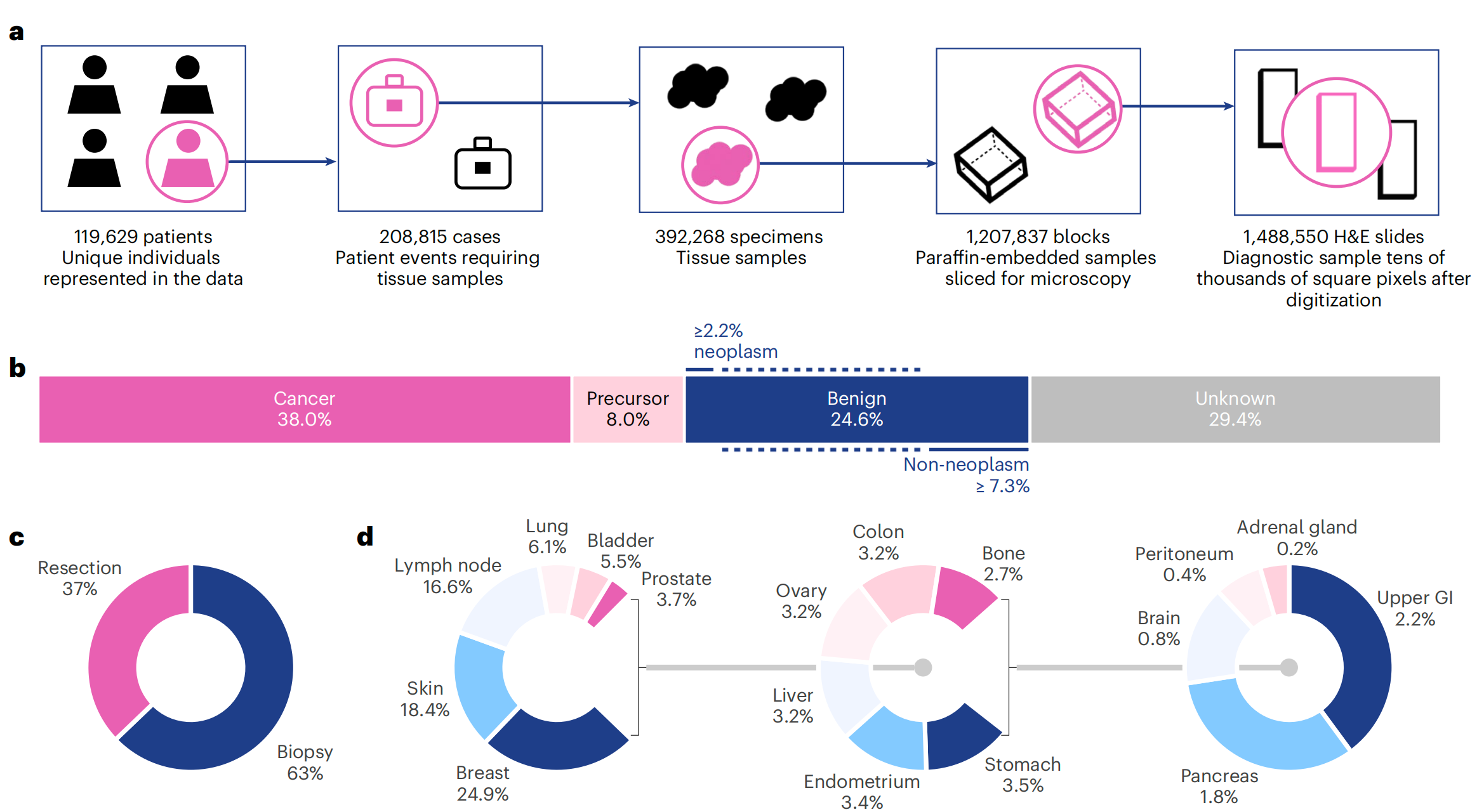
a. 训练数据描述:数据集可以从多个维度描述,包括患者(patients)、病例(cases)、样本(specimens)、组织块(blocks)或切片(slides)。这些维度展示了数据集的规模和多样性。
b. 癌症状态的切片分布:这部分展示了不同癌症状态下的切片分布,可能包括癌症、前体病变(如原位癌)、良性病变和未知状态。这有助于理解模型在不同癌症类型上的表现。
c. 手术类型的切片分布:这部分展示了根据手术类型(如活检、切除等)的切片分布。这有助于评估模型在不同手术背景下的适用性。
d. 组织类型的切片分布:这部分展示了不同组织类型的切片分布,如乳腺、皮肤、淋巴结等。这有助于了解模型在不同组织类型上的泛化能力。
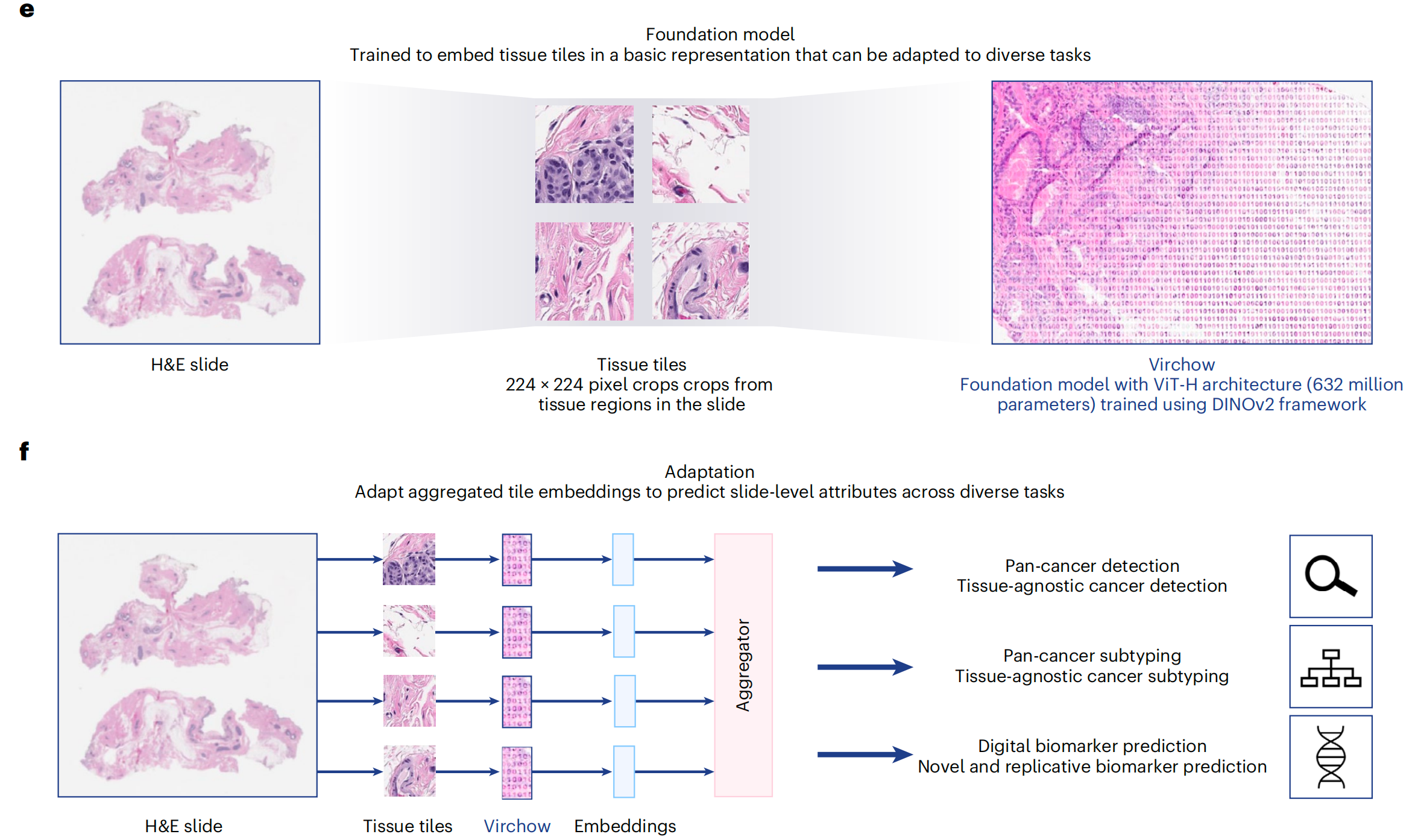
e. 训练中的数据流:在训练过程中,切片被处理成小块(tiles),然后这些小块被裁剪成全局视图和局部视图。这种处理方式有助于模型学习到更丰富的局部和全局特征。
f. 基础模型的应用:展示了如何使用聚合器模型(aggregator model)来预测切片级别的属性。聚合器模型将小块嵌入(tile embeddings)聚合起来,以预测整个切片的属性,如癌症存在与否、癌症类型等。GI(胃肠道)在这里可能是指模型在胃肠道相关病理图像上的应用。
总结来说,图1展示了Virchow模型从训练数据的准备到模型训练,再到最终应用的整个流程。通过这种设计,Virchow能够处理和分析大规模的病理图像数据,为临床病理学提供支持。
二、发现肝细胞癌治疗新靶点:脂质代谢与自噬的交叉作用

一作&通讯
| 角色 | 姓名 | 单位(中文) |
|---|---|---|
| 第一作者 | Qingbin Liu | 济宁市第一人民医院临床医学院实验中心,山东省第一医科大学,中国 |
| 并列第一作者 | Xiangyu Zhang | 济宁市第一人民医院病理学系,山东省第一医科大学,中国 |
| 并列第一作者 | Jingjing Qi | 奥地利林茨约翰尼斯开普勒大学医学院,奥地利 |
| 通讯作者 | Shulong Jiang | 济宁市第一人民医院临床医学院实验中心,山东省第一医科大学,中国 |
| 通讯作者 | Yong Yu | 奥地利林茨约翰尼斯开普勒大学医学院,奥地利 |
| 通讯作者 | Changlin Ma | 济宁市第一人民医院肝胆外科,山东省第一医科大学,中国 |
文献概述
这篇文章通过综合分析HCC的脂质代谢重编程,揭示了其在精准医疗中的潜在应用,并识别了新的预后标志物和治疗靶点。
研究背景与目的:
- HCC是全球癌症相关死亡的第二大原因,具有高度的异质性,由多种遗传变异驱动,导致缺乏有效的治疗选择。
- 本研究通过系统的多组学特征分析HCC,以揭示其代谢重编程的签名。
方法与结果:
- 通过整合转录组学、代谢组学和脂质组学的综合分析,研究者发现了HCC中与葡萄糖通量、脂质氧化降解和新合成脂肪酸有关的代谢途径的显著变化。
- 脂质组学分析揭示了甘油酯、磷脂酰胆碱和鞘脂衍生物的异常变化。
- 利用机器学习技术,研究者识别了与脂质代谢相关的基因组,作为不同病因HCC的共同生物标志物。
- 研究表明,靶向磷脂酰胆碱(PC)的饱和脂肪酸和长链鞘脂生物合成途径,特别是通过抑制溶血磷脂酰胆碱酰转移酶1(LPCAT1)和神经酰胺合酶5(CERS5),可能是HCC治疗的潜在策略。
- 数据还揭示了CERS5在促进肿瘤进展中的致癌作用,特别是通过脂质自噬。
结论:
- 研究阐明了HCC中脂质代谢的重编程特性,识别了预后标志物和治疗靶点,并强调了HCC治疗干预中潜在的与代谢相关的靶点。
文章还包括了对HCC的转录组、代谢组和脂质组的全面分析,以及通过机器学习方法鉴定HCC的代谢诊断生物标志物。此外,研究还探讨了CERS5在HCC中的作用,以及通过靶向脂质代谢途径对HCC进行潜在治疗的策略。
知识点补充:脂质代谢重编程
脂质代谢重编程(Lipid Metabolic Reprogramming)是指在某些生理或病理状态下,细胞或组织对脂质的合成、分解、转运和信号传导等代谢途径进行调整的过程。
这种代谢调整通常是为了满足细胞在特定环境下的能量需求、结构需求或信号传导需求。在肿瘤生物学中,脂质代谢重编程尤为重要,因为它与肿瘤细胞的生存、增殖、侵袭和转移等能力密切相关。
在肿瘤细胞中,脂质代谢重编程包括以下几个方面:
- 增强的脂肪酸合成:肿瘤细胞可能会增加脂肪酸的合成,以满足细胞膜的生物合成需求和能量存储。
- 脂肪酸氧化的改变:肿瘤细胞可能会调整脂肪酸的氧化过程,以适应快速增殖的能量需求。
- 脂质储存的变化:肿瘤细胞可能会增加或减少脂质储存,如三酰甘油(TG)和胆固醇酯,以适应不同的代谢状态。
- 脂质去饱和酶的活性改变:通过改变脂质去饱和酶的活性,肿瘤细胞可以调节膜脂的流动性和信号传导。
- 鞘脂类代谢的调整:鞘脂类如神经酰胺和其衍生物在细胞信号传导、细胞死亡和细胞存活中扮演重要角色,肿瘤细胞可能会调整这些代谢途径来促进其生存和增殖。
- 脂质介导的信号通路的改变:肿瘤细胞可能会利用脂质作为信号分子,通过激活或抑制特定的信号通路来促进肿瘤发展。
在HCC(肝细胞癌)的背景下,脂质代谢重编程可能涉及到上述某些或全部方面,并且可能与肿瘤的异质性、进展和对治疗的响应有关。通过深入理解这些变化,研究人员可以发现新的生物标志物和治疗靶点,从而提高HCC的诊断、预后评估和治疗效果。
重点关注
FIGURE 1 提供了肝细胞癌(HCC)中代谢组、脂质组和转录组分析的概况。
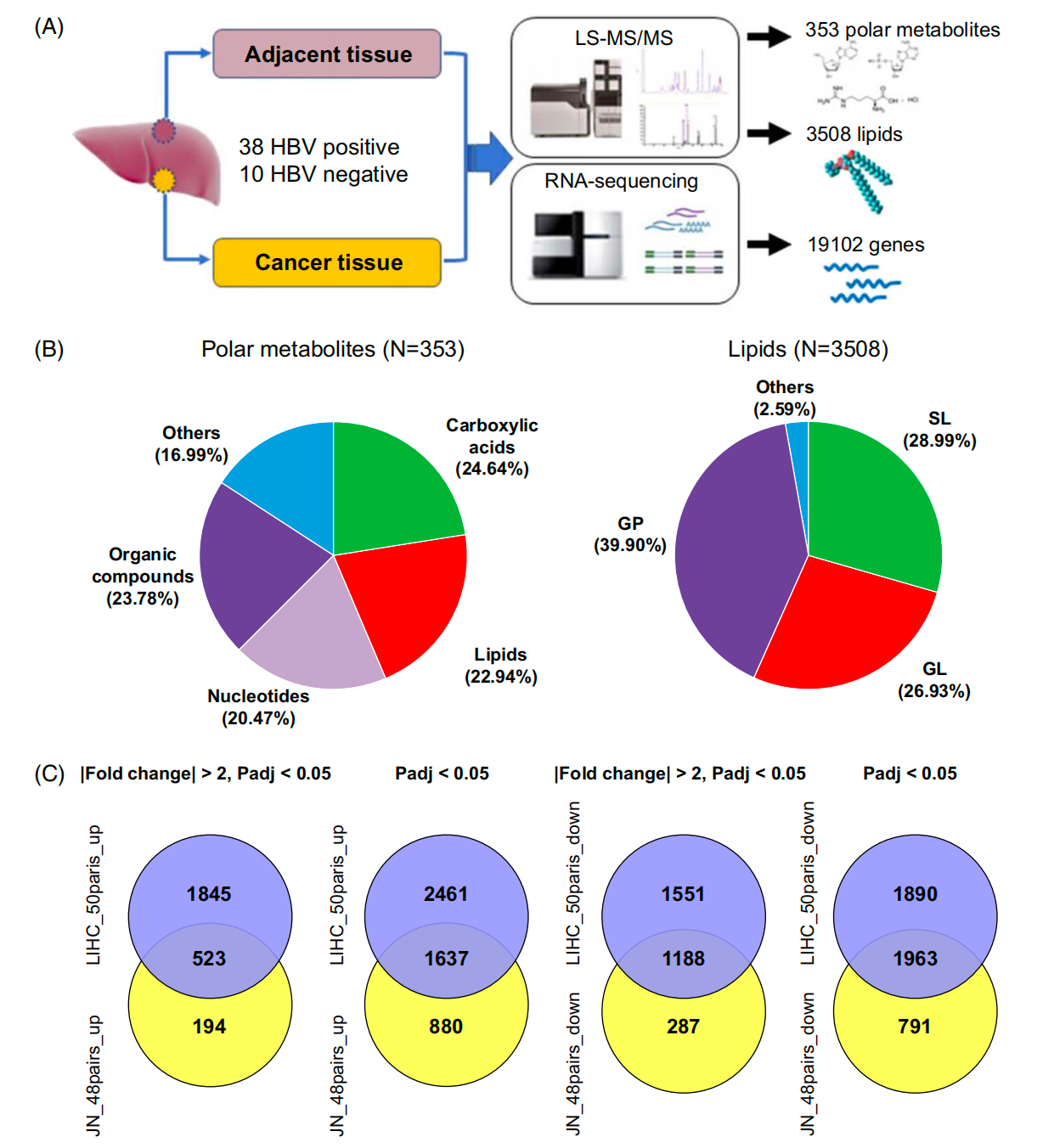
(A) 多组学方法的示意图:
- 这部分提供了一个示意图,展示了通过多组学方法识别样本中的分子。
- 它包括了实验设计、样本收集、分析流程以及不同组学层次的数据如何整合来提供对HCC分子特征的全面理解。
(B) 已注释代谢物和脂质类别的数量和比例:
- 这部分展示了研究中鉴定出的极性代谢物和脂质的数量,并以饼状图的形式表示了它们在总脂质中所占的比例。
- 饼状图详细列出了不同类别的脂质,如甘油酯、磷脂酰胆碱(PC)、鞘脂(SL)等,以及它们在HCC中的相对丰度。
- 这有助于识别HCC中哪些脂质途径可能发生改变,为进一步的生物标志物发现和机制研究提供线索。
© JN_48pairs队列与TCGA LIHC_50pairs队列中差异表达基因(DEGs)的比较:
- 这部分比较了两个不同队列(JN_48pairs和TCGA LIHC_50pairs)之间差异表达基因的情况。
- 差异表达基因是指在不同组织或条件下表达水平有显著变化的基因,它们可能与HCC的发生发展密切相关。
- 通过比较,研究人员可以识别两个数据集之间的一致性和差异性,这有助于验证生物标志物的稳健性,并揭示可能影响HCC异质性的遗传背景和环境因素。
整体来看,FIGURE 1 为理解HCC在代谢和脂质层面的变化提供了一个宏观视角,并为后续的生物信息学分析和生物学验证奠定了基础。
三、数字化端粒测量技术揭示健康老化与疾病的差异

一作&通讯
| 作者角色 | 作者姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Santiago E. Sanchez | 斯坦福大学医学院,斯坦福癌症研究所,斯坦福,加利福尼亚,美国 |
| 通讯作者 | Steven E. Artandi | 斯坦福大学医学院,斯坦福癌症研究所,斯坦福,加利福尼亚,美国 |
文献概述
这篇文章报道了一种新的数字化端粒测量技术,通过长读序列区分健康老化与疾病,为理解端粒在老化和疾病中的作用提供了新的视角。
-
端粒长度的重要性:端粒长度是生物体老化和细胞复制潜力的重要生物标志物,但现有的测量方法在分辨率和准确性上存在限制。
-
数字端粒测量技术:作者使用基于纳米孔测序的DTM技术,以高达30个碱基对的分辨率测量人类细胞中的端粒长度,并观察端粒长度的缩短和新合成的延长。
-
健康老化与疾病中的端粒变化:研究发现,随着年龄的增长,人类端粒逐渐变短,特别是长端粒的丢失和短端粒的累积。在端粒维持有遗传缺陷的患者中,短端粒的累积更为明显,并与疾病的严重程度相关。
-
机器学习的应用:研究者应用机器学习训练了一个二元分类模型,用于区分健康个体和患有端粒生物学障碍的人群。
-
端粒的生物学功能:端粒是核蛋白结构,防止染色体末端被识别为双链断裂,并招募端粒酶,这是一种在干细胞和癌细胞中负责维持端粒长度的核糖核蛋白全酶。
-
端粒缩短的机制:在没有端粒酶的非恶性细胞中,由于DNA聚合酶无法完全复制滞后DNA链,端粒在每次细胞分裂时缩短数十个碱基对。
-
端粒危机:在Rb和TP53受损的细胞中,功能失调的端粒会引发端粒危机,这是一种以缺乏功能性端粒的染色体末端发生端到端融合为特征的过程,导致染色体不稳定。
-
端粒酶的作用:在自我更新的细胞中,如干细胞、生殖细胞和癌细胞,端粒酶通过直接延长端粒来抵消末端复制问题。
-
研究方法:研究者开发了一个名为Telometer的测序准备和生物信息学流程,能够从全基因组或端粒富集的长读中重复测量端粒。
-
研究结果:通过长读序列测量的端粒平均长度与现有的金标准(TRF Southern blot和flow-FISH)高度相关。通过自举分析,测量方法的标准误差随着额外端粒测量的数量呈指数衰减,最终达到30-40个碱基对的最大精度。
-
临床应用潜力:DTM技术有望作为评估老化和疾病的临床生物标志物,特别是在区分健康老化与端粒生物学障碍(TBDs)方面。
文章还包括了详细的实验方法、数据分析、二元分类模型的构建和验证,以及对研究结果的深入讨论。
重点关注
Fig. 1 展示了通过纳米孔长读序列进行高分辨率端粒测量的一系列实验结果。
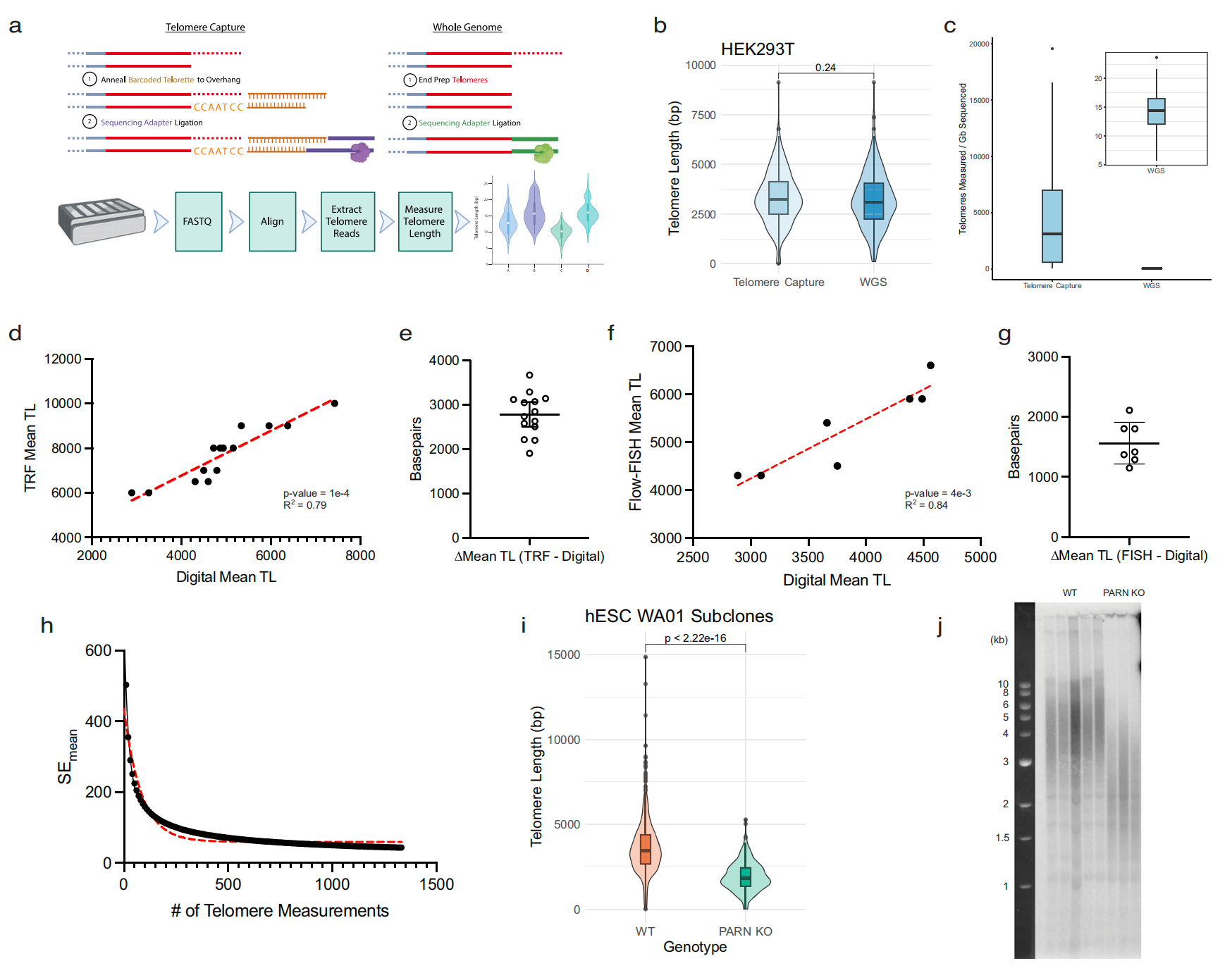
a. 示意图:说明了用于端粒测量的DNA测序文库的准备方法,包括端粒捕获或全基因组长读序列。
b. 端粒长度分布的直接比较:对比了通过端粒捕获(417个端粒测量)和全基因组测序(513个端粒测量)两种文库准备方法得到的端粒长度分布。这些数据来源于单一来源的HEK 293 T DNA。
c. 每吉咖碱基序列的端粒数:展示了两种文库准备方法的端粒测序效率(14次端粒捕获实验;23次全基因组测序)。
d. 相关性分析:显示了通过测序或TRF(Southern blot)确定的匹配样本的平均端粒长度之间的相关性(14个样本,p值为1e-4,R²=0.79)。
e. 差异分析:显示了通过TRF和测序方法测量的匹配样本的平均端粒长度之间的碱基对差异(14个个体样本,用实线和误差条表示均值±标准差)。
f. RTEL1突变个体的相关性分析:展示了通过测序或流式-FISH(flow-FISH)确定的RTEL1突变个体样本的平均端粒长度之间的相关性(7个样本,p值为4e-3,R²=0.84)。
g. 流式-FISH与测序的差异分析:显示了通过流式-FISH和测序方法测量的匹配样本的平均端粒长度之间的碱基对差异(7个个体样本)。
h. 自举分析:分析了通过迭代随机抽样替换测量值来计算平均端粒长度的标准误差的变化。
i. 数字端粒测量:展示了野生型(1333个端粒,橙色)和PARN基因敲除的hESCs(407个端粒,绿色)的测序结果。
j. 模拟端粒测量:通过TRF方法对野生型(5个独立亚克隆)和PARN基因敲除的hESC亚克隆(3个独立亚克隆)进行的端粒长度测量。
这些图表和分析结果表明,长读序列技术在测量端粒长度方面具有高分辨率和准确性,与传统的TRF和流式-FISH方法相比,提供了更多的信息,并且能够检测到端粒长度的细微变化。
四、不宁腿综合征风险预测与药物靶点发现的基因组学进展

一作&通讯
| 角色 | 作者姓名 | 单位名称(中文翻译) |
|---|---|---|
| 第一作者 | Barbara Schormair | 德国环境健康研究中心,神经基因组学研究所 |
| 通讯作者1 | Juliane Winkelmann | 德国环境健康研究中心,神经基因组学研究所 |
| 通讯作者2 | Emanuele Di Angelantonio | 剑桥大学,英国心脏基金会心血管流行病学研究部,剑桥大学公共卫生和初级保健系 |
| 通讯作者3 | Konrad Oexle | 德国环境健康研究中心,神经基因组学研究所 |
文献概述
这篇文章通过全基因组关联研究的元分析,揭示了不宁腿综合征的遗传结构,为疾病生物学的理解、风险预测和潜在治疗靶点的发现提供了重要见解。
-
研究背景:不宁腿综合征(RLS)是一种影响高达10%老年人口的慢性感觉运动障碍,其诊断和治疗常常被延误。该研究旨在通过全基因组关联研究(GWAS)的元分析来推进疾病预测并寻找新的治疗方法。
-
研究方法:研究者在116,647名RLS患者和1,546,466名欧洲血统的对照个体中进行了全基因组关联研究的元分析。研究还包括性别特异性的GWAS和X染色体的遗传研究。
-
主要发现:
- 通过汇总分析,风险位点的数量增加了八倍,达到164个,包括三个在X染色体上的位点。
- 性别特异性的元分析显示,两性之间的遗传易感性高度重叠(rg = 0.96)。
- 基因注释优先考虑了可药物作用的基因,如谷氨酸受体1和4。
- 孟德尔随机化(Mendelian randomization)表明RLS是糖尿病的一个因果风险因素。
- 结合遗传和非遗传信息的机器学习方法在风险预测中表现最佳(AUC = 0.82–0.91)。
-
功能注释和生物学解释:
- 通过基因集和细胞类型富集分析,研究者识别了与RLS相关的神经发育、神经元迁移、轴突导向、突触形成和神经元间信号传导的途径。
- 在小鼠和人类发育阶段的单细胞测序数据中,神经元和神经母细胞显示出显著的富集。
-
药物再利用:研究者根据基因注释和药物基因组学,识别了13个潜在的药物再利用目标,包括编码AMPA型谷氨酸离子型受体亚基的GRIA1和GRIA4。
-
遗传相关性和孟德尔随机化分析:通过大规模遗传相关性分析和孟德尔随机化,研究者发现了RLS与其他疾病(如失眠、抑郁、高血压、心血管疾病、糖尿病和睡眠障碍)之间的关联,并探索了这些关联的因果关系。
-
风险预测模型的开发和验证:研究者评估了基于遗传数据和基本人口统计变量(如年龄、性别和疾病发病年龄)的预测模型。机器学习生存分类器模型在考虑非线性相互作用和时间变化效应时表现最佳。
-
讨论:文章讨论了研究的局限性,包括缺乏详细的临床数据和非欧洲人群的GWAS研究,并提出了未来研究的方向。
重点关注
图1展示了通过途径富集分析得到的显著富集的途径(FDR < 0.05)。分析使用了两种不同的方法:DEPICT和MAGMA。

DEPICT分析:
- 使用单样本单侧Z检验。
- 将基因本体(GO)术语基于语义相似性进行聚类,使用的是Wang的GOSemSim方法,该方法在rrvgo软件包1.2.0版本中实现。
- 术语以矩形呈现,颜色表示术语属于特定聚类。
- 每个聚类通过粗边框线可视化。
- 矩形的大小对应于富集的显著性,每个聚类中最显著富集的术语被选为代表性术语,并以白色字体显示。
MAGMA分析:
- 使用单侧t检验。
- 与DEPICT类似,MAGMA分析也基于语义相似性对GO术语进行聚类,但具体方法可能有所不同。
- 术语同样以矩形呈现,颜色和边框线的使用方式与DEPICT相同。
总结:
- 这两种方法都用于识别在不宁腿综合征(RLS)中显著富集的生物学途径。
- 聚类和可视化帮助理解不同途径之间的关联以及它们在RLS生物学中可能的作用。
- 显著富集的途径可能为理解RLS的分子机制和开发新的治疗方法提供线索。
五、多模态人工智能生物标志物在高风险前列腺癌预后评估中的验证

一作&通讯
| 作者角色 | 作者姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Daniel E. Spratt | 凯斯西储大学西德曼癌症中心,克利夫兰,俄亥俄州,美国 |
文献概述
这篇文章通过元分析六个NRG/RTOG第3阶段随机试验的个体患者数据,验证了多模态人工智能生物标志物ArteraAI Prostate在预测高风险局部前列腺癌患者远处转移、前列腺癌特异性死亡率和带有远处转移的死亡方面的独立预后能力。
研究收集了来自NRG/RTOG生物库的六项试验的组织病理学切片,并将它们数字化。这些试验包括原始MMAI验证队列中使用的四项随机试验(NRG/RTOG 9202, 9408, 9413, 和 9910; n = 426)以及两项未参与初始MMAI开发和验证过程的独立试验(NRG/RTOG 0521, n = 344; NRG/RTOG 9902, n = 318)。将数字化图像与临床信息结合,为每位患者生成了一个MMAI得分。
研究使用Fine-Gray和累积发生率分析来评估远处转移(DM)、前列腺癌特异性死亡率(PCSM)和带有远处转移的死亡(DDM)的时间,并根据MMAI模型得分进行分层,既作为连续得分(每标准差的增量)也按四分位数进行分类。多变量分析(MVAs)用于展示MMAI模型的独立效应和NCCN H/VH风险因素的数量。
研究结果显示,即使患者至少有一个NCCN H/VH风险因素,MMAI生物标志物也能识别出具有高度可变风险的DM、PCSM和DDM患者。MMAI模型在调整了H/VH风险特征数量后,仍然显著地与DM、PCSM和DDM相关联。10年的DM风险估计值在MMAI四分位数Q1-2为8%,而在MMAI Q3-4为26%,绝对差异为18%。
这项研究验证了MMAI生物标志物在六个第3阶段随机试验中的高风险前列腺癌患者数据中的有效性,并独立于标准临床和病理变量。研究还提出了未来工作可能扩展到预后之外,通过评估MMAI生物标志物与特定治疗的相互作用,为医生和患者提供更个性化的治疗决策信息。
重点关注
Fig. 1 包含了四个部分,分别展示了MMAI得分与高风险局部前列腺癌患者的不同临床结果之间的关系。
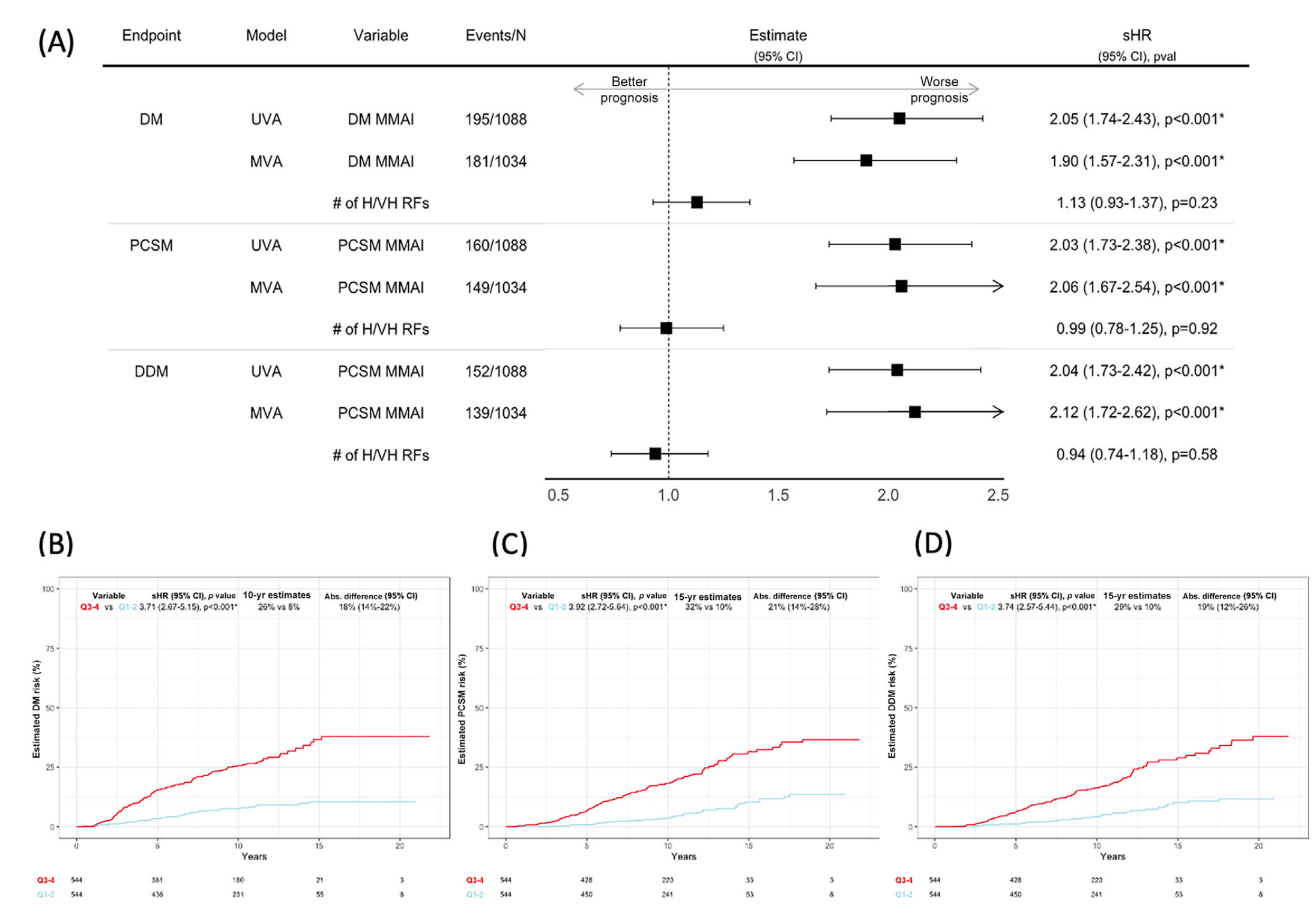
-
(A) UVA of the association between the MMAI score and DM and MVA of the association between the MMAI score and DM after adjustment for the number of H/VH risk factors.
- 这部分展示了MMAI得分与远处转移(DM)之间的关联。首先进行了单变量分析(UVA),然后进行了多变量分析(MVA),在MVA中调整了高/非常高(H/VH)风险因素的数量。这表明了即使在考虑了其他风险因素后,MMAI得分仍然是一个独立的预后因素。
-
(B) Estimated 10-yr DM rates for MMAI Q1–2 versus Q3–4.
- 这部分估计了MMAI得分在第一和第二四分位数(Q1-Q2)与第三和第四四分位数(Q3-Q4)的患者之间,10年远处转移率的差异。这可能显示了MMAI得分较低的患者与得分较高的患者相比,远处转移的风险有显著差异。
-
© Estimated 15-yr PCSM rates for MMAI Q1–2 versus Q3–4.
- 这部分与第2点类似,但关注的是15年前列腺癌特异性死亡率(PCSM)的估计率。它可能揭示了MMAI得分较低的患者与得分较高的患者相比,长期生存率的差异。
-
(D) Estimated 15-yr DDM rates for MMAI Q1–2 versus Q3–4.
- 这部分估计了15年带有远处转移的死亡(DDM)率,同样比较了MMAI得分较低与较高的患者群体。这有助于了解MMAI得分对于预测患者长期生存和远处转移风险的影响。
整体而言,Fig. 1 通过不同的统计分析方法和时间点,展示了MMAI得分如何作为一个强有力的预后工具,帮助区分高风险局部前列腺癌患者的不同临床结果风险。
六、非侵入性癌症监测:MRD-EDGE技术提升ctDNA检测灵敏度

一作&通讯
| 作者角色 | 作者姓名 | 单位名称(英文) | 单位名称(中文) |
|---|---|---|---|
| 第一作者 | Adam J. Widman | New York Genome Center, Memorial Sloan Kettering Cancer Center | 纽约基因组中心,纪念斯隆凯特琳癌症中心 |
| Weill Cornell Medicine | 威尔康奈尔医学院 | ||
| 通讯作者 | Nicolas Robine | New York Genome Center | 纽约基因组中心 |
| Claus Lindbjerg Andersen | Aarhus University Hospital, Aarhus University | 奥胡斯大学医院,奥胡斯大学 | |
| Dan A. Landau | New York Genome Center, Memorial Sloan Kettering Cancer Center, Weill Cornell Medicine | 纽约基因组中心,纪念斯隆凯特琳癌症中心,威尔康奈尔医学院 |
文献概述
利用机器学习引导的信号增强技术,MRD-EDGE能够通过血浆中的循环肿瘤DNA进行超敏感的肿瘤负担监测,为癌症治疗和监测提供了新的策略。
MRD-EDGE技术通过全基因组测序(WGS)检测ctDNA中的单核苷酸变异(SNV)和拷贝数变异(CNV),以提高信号的富集度。其中,MRD-EDGESNV利用深度学习和ctDNA特定的特征空间,将WGS中的SNV信号与噪声的富集度提高了约300倍。MRD-EDGECNV则减少了通过WGS进行超敏感CNV检测所需的非整倍体程度,从1Gb降低到200Mb,从而扩大了其在实体瘤中的适用性。
研究团队利用MRD-EDGE技术在多种癌症类型中识别手术后的MRD,追踪肺癌中新辅助免疫疗法引起的肿瘤分数(TF)变化,并在结直肠腺瘤中展示了ctDNA的脱落。此外,MRD-EDGESNV在提高信号与噪声的富集度方面取得了突破,使得在晚期黑色素瘤和肺癌中可以进行仅基于血浆的(非肿瘤信息的)疾病监测,为接受免疫检查点抑制治疗的患者提供了具有临床意义的TF监测。
文章还讨论了液体活检如何通过非侵入性检测和监测血浆中的ctDNA来重塑癌症护理,以及如何通过深度靶向测序来克服ctDNA在低肿瘤分数设置中的稀疏性。研究者们通过一系列实验验证了MRD-EDGE技术的性能,包括在黑色素瘤、非小细胞肺癌(NSCLC)和结直肠癌(CRC)中检测低TFs的ctDNA。
总之,这项研究展示了MRD-EDGE作为一种新的监测工具,能够在多种临床环境中提供对肿瘤负担的超敏感评估,具有改善癌症患者治疗和管理的潜力。
知识点补充:单细胞测序与全基因组测序的区别和联系
单细胞测序(Single-cell sequencing)和全基因组测序(Whole-genome sequencing,WGS)是两种不同的基因组学技术,它们在研究目的、技术方法和应用领域上有所区别,但也存在一定的联系。
区别:
-
样本尺度:
- 单细胞测序:专注于单个细胞的基因表达或基因组变异,能够揭示细胞间的异质性。
- 全基因组测序:对整个基因组进行测序,通常用于分析个体的完整遗传信息。
-
技术复杂性:
- 单细胞测序技术更为复杂,需要将单个细胞分离并单独进行测序。
- 全基因组测序通常涉及大量细胞的DNA,技术过程相对简单。
-
数据类型:
- 单细胞测序可以提供关于单个细胞状态的详细信息,如基因表达水平、拷贝数变异等。
- 全基因组测序提供个体的全面遗传信息,包括单核苷酸多态性(SNPs)、插入/缺失(InDels)和结构变异等。
-
应用领域:
- 单细胞测序常用于细胞发育、疾病机理、肿瘤异质性等研究。
- 全基因组测序适用于遗传病诊断、癌症基因组分析、物种进化研究等。
-
成本和通量:
- 单细胞测序成本较高,每次实验分析的细胞数量有限。
- 全基因组测序成本相对较低,可以一次性分析整个基因组。
联系:
-
遗传信息:两者都提供了关于遗传信息的数据,有助于理解基因组的结构和功能。
-
技术发展:单细胞测序技术的发展受益于全基因组测序技术的成熟,例如在测序平台和数据分析方法上。
-
研究目的:两者都可用于疾病机理的研究,尤其是在癌症基因组学中,单细胞测序可以帮助理解肿瘤内部的异质性,而全基因组测序可以揭示整个肿瘤基因组的变化。
-
数据整合:在某些研究中,单细胞测序数据和全基因组测序数据可以整合使用,以获得更全面的生物学见解。
-
技术交叉:例如,单细胞全基因组测序(single-cell whole-genome sequencing)结合了两种技术,可以同时分析单个细胞的基因组变异和基因表达。
总的来说,单细胞测序提供了细胞层面的详细视图,而全基因组测序提供了整个基因组的全面视图。两者结合使用可以为生物学和医学研究提供更深入的理解。
重点关注
图2 展示了基于机器学习的错误抑制和额外特征如何增强基于血浆全基因组测序(WGS)的拷贝数变异(CNV)检测的灵敏度。
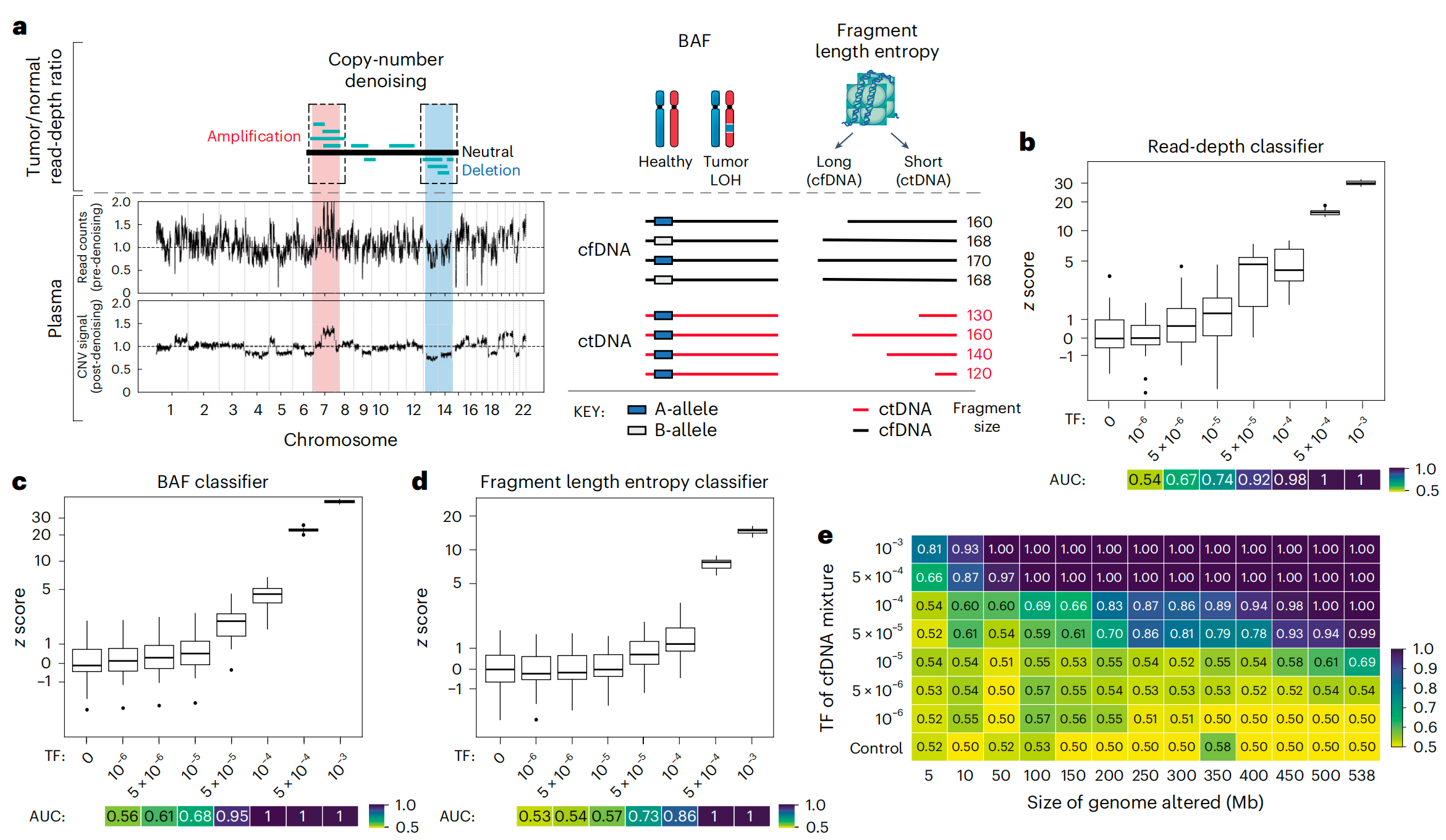
a. 拷贝数去噪以推断血浆读取深度:
- 通过比较肿瘤和正常基因组WGS,选择患者特异性的CNV片段。
- 在血浆中,CNV片段可能被原始读取深度轮廓中的噪声掩盖。
- 使用健康对照血浆样本(PON)进行机器学习引导的去噪,去除背景噪声,生成去噪后的血浆读取深度轮廓。
- PON血浆样本不包括在下游CNV分析中。
- 可以通过cfDNA中SNPs的BAF变化来测量LOH(杂合性缺失)。
- 由于ctDNA(较短的片段大小)对血浆cfDNA池的贡献不同,预计在肿瘤扩增或删除区域会增加或减少片段长度异质性。
- 通过片段插入大小的香农熵来测量片段长度异质性。
b-e. 体外混合研究:
- 对来自结直肠癌(CRC)患者的高和低肿瘤分数(TF)样本进行体外混合研究。
- 将治疗前血浆(TF = 12%)混合到非癌症血浆(CTRL-443;b,d)或匹配的PBMCs(c)中,共25个重复样本。
- 混合样本模拟了 1 0 − 6 10^{-6} 10−6到 1 0 − 3 10^{-3} 10−3的肿瘤分数。
- 箱形图表示中位数、下四分位数和上四分位数,须对应1.5倍四分位距。
- AUC热图展示了不同混合TFs与阴性对照(TF = 0)的检测性能,通过z分数测量(由读取深度偏斜的总和、BAF分类器的BAF分数和片段长度熵的总和得出)。
- 读取深度分类器在TF > 0时显示出检测灵敏度,低至5×10^-5(AUC 0.92)。
- SNP BAF(c)和片段长度熵(d)分类器在5×10^-5时显示出检测灵敏度(AUC分别为0.95和0.73)。
- 测量MRD-EDGECNV的LLOD(最低检测限)对于组合特征集,作为CNV负荷和混合模型TF的函数(e)。
- 在TF = 5×10^-5和200Mb时观察到敏感检测(AUC 0.70)。
- 控制行显示了另外25个TF = 0种子,这些种子未包括在下采样分析中。
- AUC被限制在0.50到1.00的范围内。
















 753
753

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









