







NerLTR-DTA: drug–target binding affinity prediction based on neighbor relationship and learning to rank
药靶相互作用预测一直被看作是一个二分类问题,即预测给定的药靶对直接是否存在边。亲和力值反映了药靶对直接相互作用的强度,目前大多数的预测方法都存在一定的挑战:(1)许多方法只讨论和应用了一个场景,注意力放在了药物再利用但忽略了新药物/靶标的发现;(2)许多方法不考虑药靶蛋白质(药物)的优先顺序。
在这项研究中,提出了一个NerLTR-DTA的模型,他通过利用邻居关系的相似性和共享性来提取特征,并且应用带有回归特性的排序框架来预测亲和力值和查询药物的靶标对的优先顺序。值得一提的是,该模型能够应用到不同的场景当中,包括新药物和靶标的发现。
模型项目地址:https://github.com/RUXIAOQING964914140/NerLTR-DTA
目录
引言
作者在引言部分论述了目前在药靶预测方面其他人做的一些工作,以及之前所提出的一些模型、技术或者方法;最后,作者总结前人的工作:许多现有的方法只能预测药物和蛋白质之间缺失的关系,其中一部分药物-靶标对信息还是已知的;他们没有注意到那些不存在相互作用信息的药靶对;而且,根据相互作用强度对候选药物进行排序,有助于选择少量有前途的药物-靶标对。
在这项研究中,提出了一个模型:NerLtr-DTA,首先,基于邻居药物节点(和当前药物节点之间存在确定的相似性和共享性的节点)信息提取特征;然后,将这些特征输入到LTR算法(Learn to rank)中,利用query和document(key)一对多的关系,通过调整q和d的类型来应用到不同的场景。
药物再利用、蛋白质新功能预测、新药物发现和新靶点发现
数据集
Davis et al. (2011) and KIBA (Tang et al.,2014) datasets,

数据预处理:(1)对于KIBA数据集中的亲和力值,首先所有值取反,然后将绝对值最小的值和每个负值相加。(这一步我是没明白为啥这样处理,后来作者说这是为了便于操作和比较……)
(2)对于Davis数据集中的亲和力值,按照以下公式变换: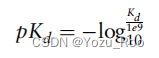
模型结构
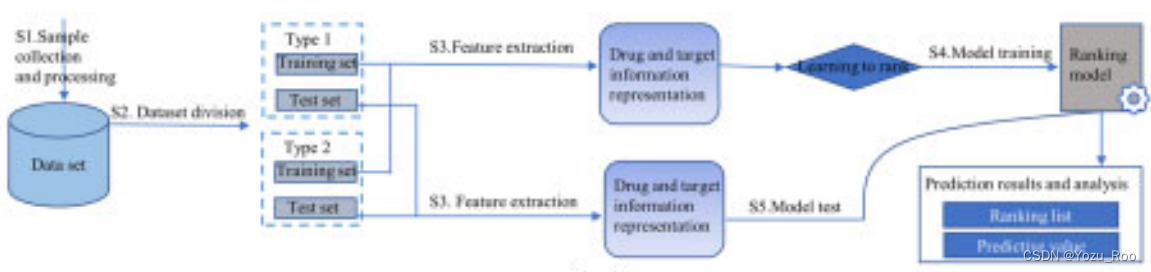
这张图在论文中就高糊,查了也没找到论文中的原图,将就着看吧,大概能看到出来整个流程的。
S1.数据的收集和处理——>S2.数据集的划分——>S3.特征提取——>药靶信息表示——>Learn to rank——>S4. 模型训练——>ranking model——>预测结果(排序列表和预测xxx 看不清了……)
S1.数据的收集和处理——>S2.数据集的划分——>S3.特征提取——>药靶信息表示——>Learn to rank——>S5. 模型测试——>ranking model——>预测结果(排序列表和预测xxx 看不清了……)
特征工程
5种类型的关联被用于药物和蛋白质的特征提取,包括:药物-药物相似性、药物-药物共享性、蛋白质-蛋白质相似性、蛋白质-蛋白质共享性和药物-蛋白质亲和力。
基于以上五种类型,提出了自关联特征(SAF)和邻近关联特征(AAF)
自关联特征
SAF来源于物体(药物或靶标)本身相关的相似性和亲和性。

邻近关联特征
AAF基于两种假设:(1)相似的药物有着相似的靶向蛋白质,反之亦然。(2)药物ds可以与一个新的靶点结合,与ds共享多个靶点的药物da很可能也可以与那个新的靶点结合。
我们利用k个与目标(药物或目标)具有高相似性和高共享性的邻居的ASAF提取AAF。此外,AAF被分为一般AAF(GAAF)和附加AAF (AAAF ).
GAAF:对于药物i(或靶标j),我们首先选择一组与药物i(或靶标j)的相似性或共享性大于规定的阈值的药物(或靶标),然后将其ASAF连接起来 将一组药物(或靶标)作为初始向量,最后将初始向量中元素的最小值、最大值、上四分位数、中位数、下四分位数、平均值和模式作为药物i(或靶标j)的GAAF。
AAAF:我们将25个额外邻居的ASAF连接为AAAF。图2d描述了另外25个邻居的选择过程。对于药物i(或靶标j),我们首先确定最多的前5个si milar药物(或靶点),这5种药物(或靶点)以O1-O5为代表,然后分别获得与O1-O5最相似的前5种药物(或靶点)。因此,我们得到了25个额外的邻居o f药物i(或目标j)。值得注意的是,药物i(或靶标j)并不包括在每种情况中。
Learning to rank (LTR)
参考资源:
排序学习(Learning to rank)综述
GBDT(MART) 迭代决策树详解‘’
本研究中作者利用了pointwise算法中的MART模型,MART作为一种具有回归属性的点态算法,不仅可以对每个查询下的相关样本进行排序,还可以尽可能地拟合预测值和实际值。
输入到LTR模型的数据格式为[affinity, qid:id, feature],每个属性的含义和表示如下:
affinity:药靶对的亲和力值;在这个研究中,K_d, K_i, IC_50用于亲和力值。
qid:id :表示查询id和与之相关的所有药物(蛋白质);例如,药靶对(Dn-T1,Dn-T2, Dn-T3,…,Dn-Tx)表示药物Dn所有的靶标对,qid:n。
featrue:是药靶的特征,我们将药靶对SAF和AAF拼接之后表示feature。
评判标准
作者选择了Concordance Index (CI), Mean Square Error (MSE) and r2_m(test)来衡量NerLTR-DTA模型的表现
-
CI的计算与含义
CI测量两个随机药物靶对的预测顺序是否与它们的真实顺序相同,其中预测顺序和真实顺序是通过比较预测和真实仿射来确定的。
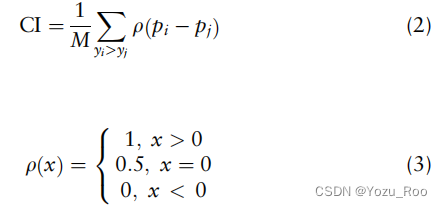
其中M是一个标准化常数。yi和yj分别表示较大和较小的亲和度值。pi和pj分别表示yi和yj的预测值。 -
MSE的计算公式和含义

其中,P和Y分别包含n个数据点的预测值和实值。Pi和Yi分别表示第i个样本的预测值和实值。
DTA:drug–target binding affinity(药靶亲和力)
- r2_m(test)的计算公式和含义
r2_m(test)是评价外部预测性能和决定QSAR模型可接受性的外部验证参数。它决定了真实值与测试样本的预测值的偏差程度。
当测试集的r2_m(test)值大于0.5时,可以确定一个可接受的、稳健的、非偶然获得的模型。

其中,r2为相关系数的平方,r2 0为截距为零的相关系数的平方。
实验与结果
多应用场景的设置策略
在这项研究中,通过设置不同的查询和抽样来应用到不同的场景中,根据对查询id的设置和训练集测试集的划分,可被分为两种策略:(1)能够预测与已知靶标(药物)结合的药物(靶标)的场景,包括药物的再利用和蛋白质新功能的预测。(2)能够预测药物(靶标)与新靶标(药物)结合的场景,包括新药物和新靶标的发现。
以上两种策略所对应的数据集的划分如下图:
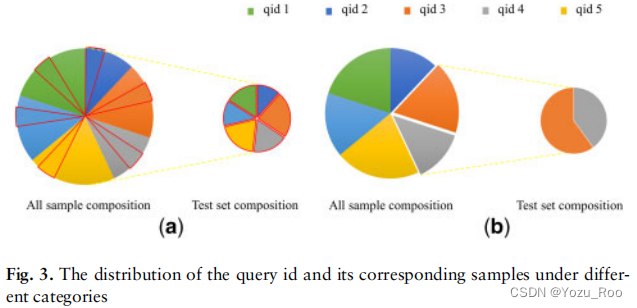
在这项研究中,在训练集中出现的药物和靶点被称为已知药物和已知靶点。否则,就被称为新药和新目标。
药物再利用:它的目的是识别已知药物潜在未发现的靶点,因此每种药物都可以被设置成一个查询。
后面就是通过设置不同的场景得到的不同的模型在不同参数下的实验数据……这里就不在过分展示和解释了,原文中都有!!!!
结论
通过和不同的模型进行比较,NerLTR-DTA有着如下的优点:
(1)模型中利用了邻居信息,使得能够更加高效的描述药靶对信息,对构造高表现力的模型十分重要
(2)模型通过设置不同query和数据集不同的划分方式来实现多场景应用,排序框架对新药物和靶标的发现十分适用。
(3)该模型是基于MART算法,一种带有回归特性的排序框架。这不仅能精准地预测具体的亲和力值,同时也能提供蛋白质的优先顺序
















 1545
1545











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











