






一.SubMDTA: drug target affinity prediction based on substructure extraction and multi-scale features
基于亚结构提取和多尺度特征的药物靶点亲和力预测 2023.9 三区
1.模型
提出了一种基于分子子结构的自监督学习方法进行分子表征。在预训练阶段,生成子图以获取子结构信息,并根据子图的相似性关系替换子图以生成重构图。同时最大化子图和原始图之间,以及重建的图和原始图之间的相关性,以提高子图级和图级表示之间的相关性。预训练后,训练后的模型在下游任务中进行微调。针对蛋白质,该文提出一种基于n-gram方法的多尺度信息整合BiLSTM方法进行特征提取。最后,将药物和蛋白质特征连接起来,并输入到多层感知器(MLP)中,用于DTA预测。
1.对于药物:SMILES字符串转换为分子图,然后使用GIN网络(GIN网络使用了全局特征聚合的思想,将节点和边的信息进行聚合得到整个图的表示。具体而言,GIN网络通过对每个节点的邻居节点特征进行汇总和聚合,然后将聚合结果与自身节点特征进行组合。这样的迭代过程可以增强节点特征的表达能力,从而更好地捕捉图的结构信息。)对图表示进行编码。GIN编码器采用多层GIN网络可以得到多个范围的局部信息,接着使用卷积层来聚合得到的多层信息来避免缺失节点信息。为了增强自监督学习特征的泛化,从ZINC数据库中随机抽取50,000个分子进行自监督模型的预训练,通过学习未标记分子数据中丰富的分子结构和语义信息,得到高质量的分子编码器。
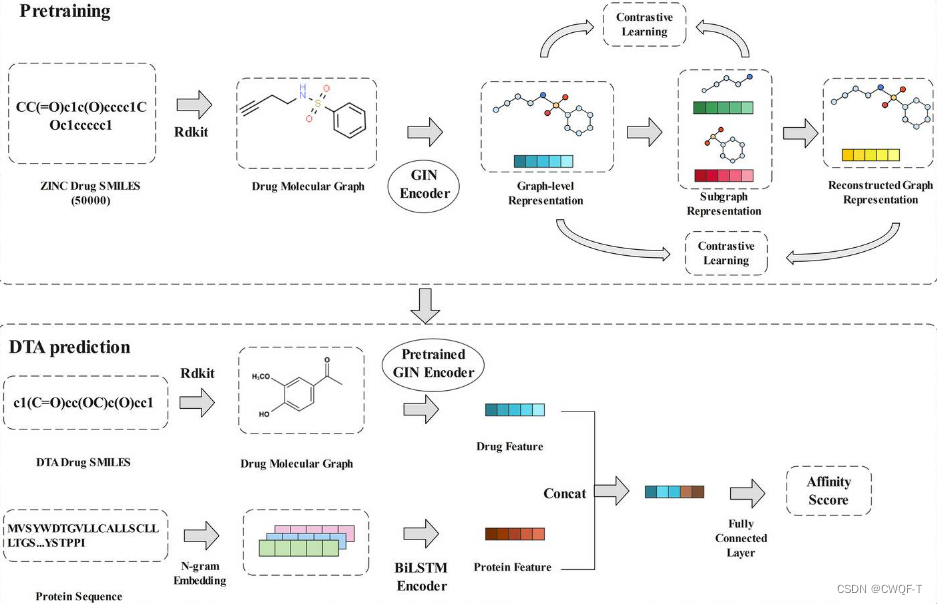
然后提取子结构图和重建图。在获得两类特征后,使用对比学习使得它们与原始图之间的互信息最大化。对比学习过程如下。对于原始图,选择其原始特征,与每个子图表示形成正样本对,与同批次中其他图的子图形成负样本对。为了捕捉图之间的内在关系,通过计算某个原始图的每个子图与其他原始图生成的子图之间的余弦相似度,可以获得一个相似度矩阵或者相似度列表,替换掉排名较低的一半的子图,以获得更好的重构图。原始图及其重构图构成正样本,而与在同一批次中的其他的重构图中构成负样本。

2.对于蛋白质序列:表示成由22个独特氨基酸组成的ASCII字符串。首先通过n-gram编码嵌入(多个n-gram),然后输入BiLSTM以获得其表征。最后,将药物表征和蛋白质表征连接并送入全连接层,以预测结合亲和力。
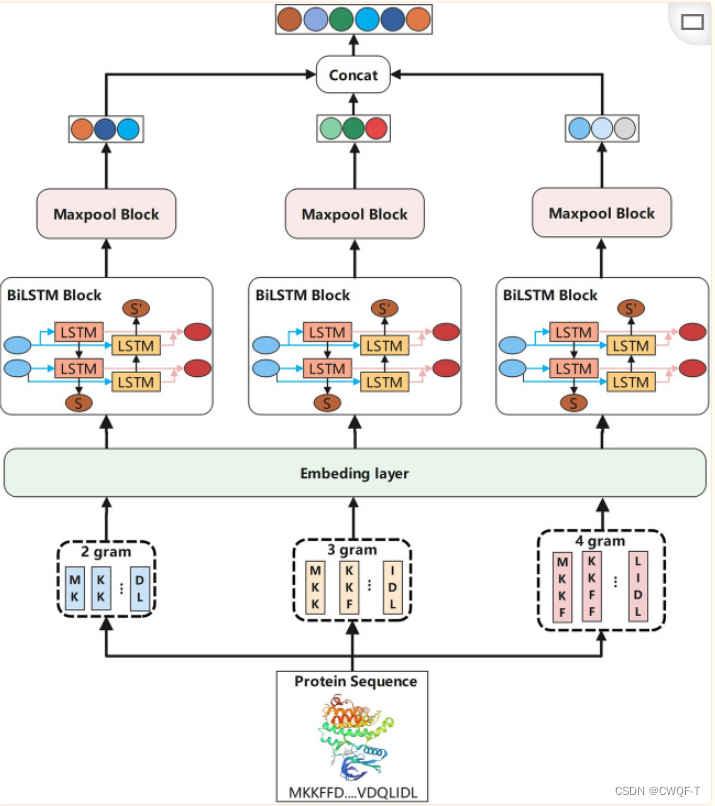
DTA 预测部分将蛋白质向量和药物向量连接起来,使用两层FC层进行预测。
1.药物表示
分子图
2.蛋白质表示
氨基酸序列
2.实验
1.数据集

3.结果
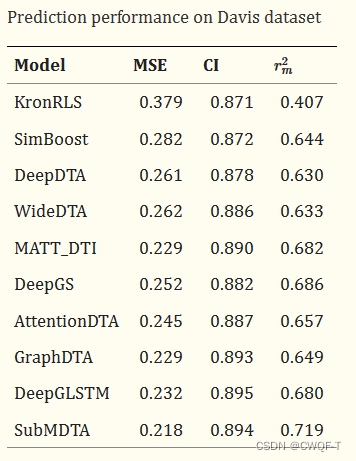
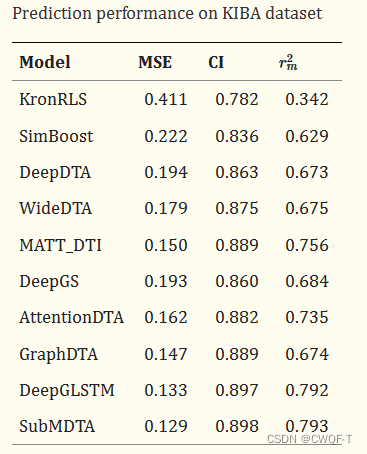
对比实验:
1.药物:与 用one-hot编码根据原子属性得到药物分子的特征、再使用GNN的分子图和分子指纹分别代替使用GIN处理分子图来进行对比。
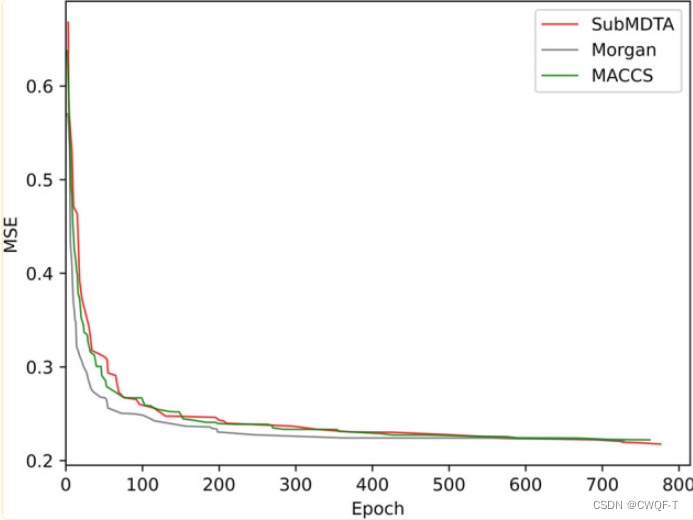
2.蛋白质:选择卷积神经网络(CNN)和双向门控循环单元(BiGRU)作为比较方法。
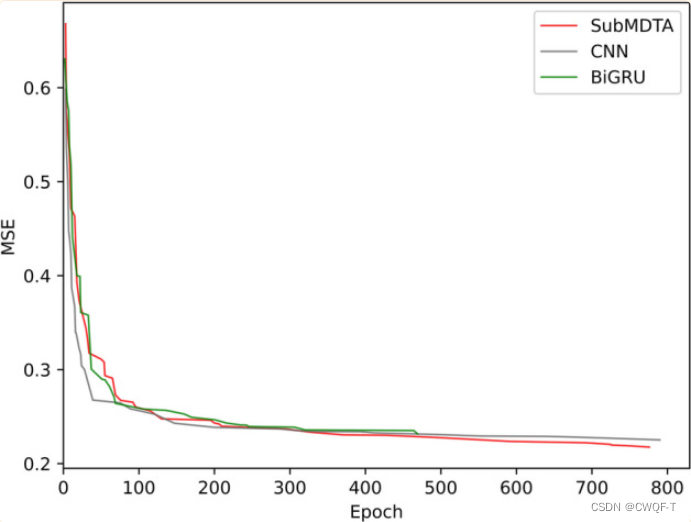
消融实验:分别与使用一种、两种对比学习以及不进行预训练进行对比

二.GEFormerDTA: drug target affinity prediction based on transformer graph for early fusion
药物靶点亲和力预测早期融合 2024.3 一区
1.模型
GEFormerDTA是一种集成了药物和蛋白质结构信息的新型神经网络模型。它利用药物四种形式的特征表示(节点、度中心、空间和边缘编码特征)来有效地利用它们在图任务中的角色。结合目标蛋白的二级结构信息和ASA信息(ASA(Accessible Surface Area)信息是指蛋白质中每个氨基酸残基暴露在溶液中的表面积),实现蛋白质结构信息的综合利用。采用早期融合机制处理药物与蛋白质之间的结合亲和力,减少信息冗余导致的预测误差。
1.对于药物来说:
节点特征:每个原子都是九个特征组成的向量。
度心特征:当一个原子与其他原子连接越多时,说明这个原子越重要。所以使用原子度中心性来表征分子图中的度特征,作为神经网络的附加信息。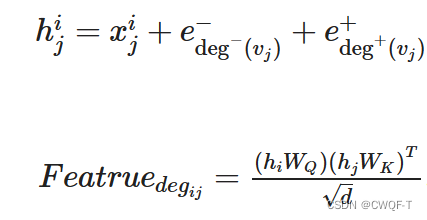
其中两个e表示分子i中的原子j的入度和初度。把原子特征h传到注意力机制中可以得到某个分子的特征。
药物空间结构信息矩阵:定义下述函数作为分子中两个原子节点之间的最短连接通路
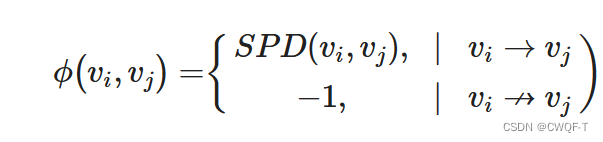
通过对度心和空间位置进行编码之后,得到了原子对(节点对)的嵌入矩阵如下,这也是原子对的空间结构特征的嵌入。
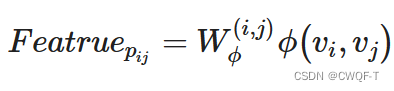
原子间的边的特征:相邻原子对之间的边被编码为如下:
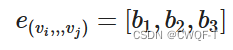 b1代表键的类型,b2代表空间键,b3代表是否共轭。都可以通过rdkit工具包获得。若ij之间最短路径是P,那这条边的特征即被定义为:
b1代表键的类型,b2代表空间键,b3代表是否共轭。都可以通过rdkit工具包获得。若ij之间最短路径是P,那这条边的特征即被定义为:
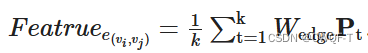
最终药物序列使用如下注意力模块加上卷积块来得到特征:
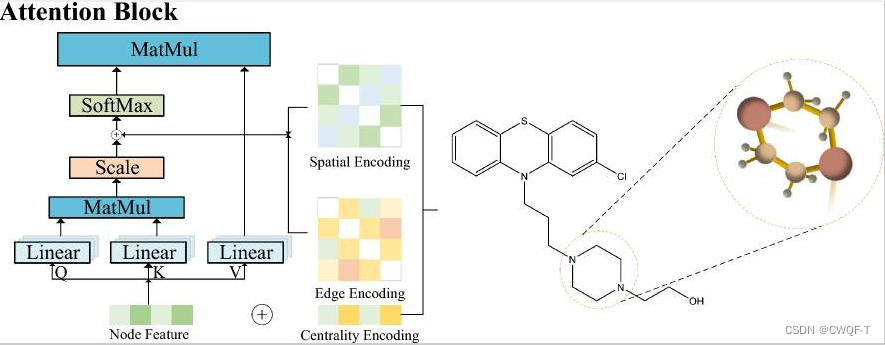
最终的药物特征还包括从分子图得到的特征
2.对于蛋白质来说:
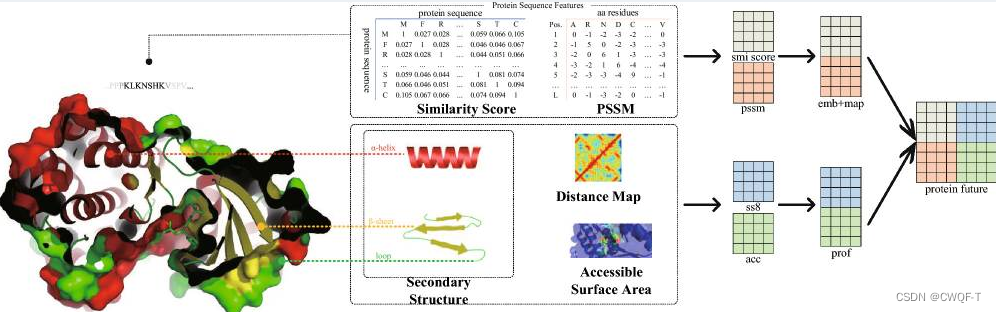
采用了 SS(相似性分数) 和 ASA用于表示蛋白质图结构。SS决定了靶蛋白的骨架结构,而ASA则表示氨基酸残基在其三维结构中与溶剂的接触或暴露程度。由肽链卷曲折叠形成的蛋白质二级结构包含有关蛋白质活性、功能和稳定性的重要信息,有利于模型预测。PSSM矩阵的每一列代表蛋白质序列中的一个位置,而每一行则代表在这个位置上可能出现的氨基酸残基。PSSM矩阵中的每个元素表示在特定位置上,某个氨基酸残基出现的频率或得分。距离图描述蛋白质内部残基之间距离关系的图示或矩阵。它展示了蛋白质中各个残基之间的空间位置和相对距离。
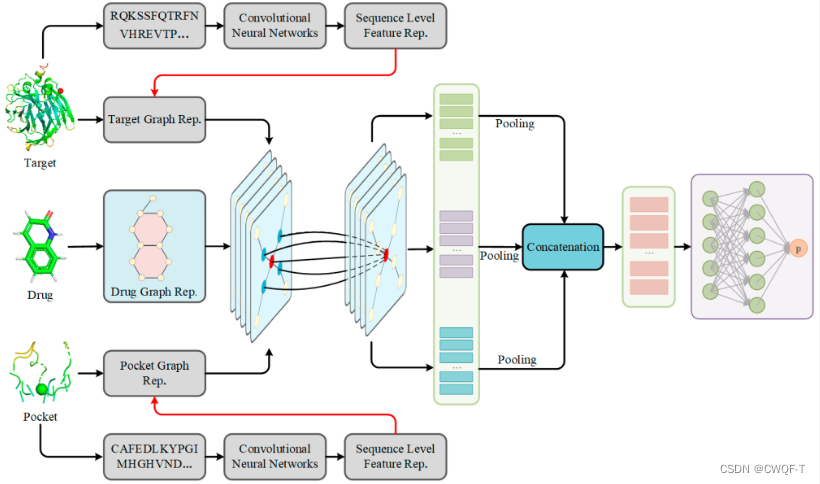
最终模型图:(a)是所提模型的数据预处理阶段。(b)是药物ESC的编码器。(c)是药物图的编码器。(d)是提出的早期融合过程的图特征。(e)是药物-靶点蛋白质图的细化过程。(f) 是DTA 最终预测过程。
在ESC模型中使用Sparsepro自注意力分子图编码器来提取重要的Q并降低模型复杂度。
在特征融合的信息池中的图将利用 GCN 来捕获基本特征信息。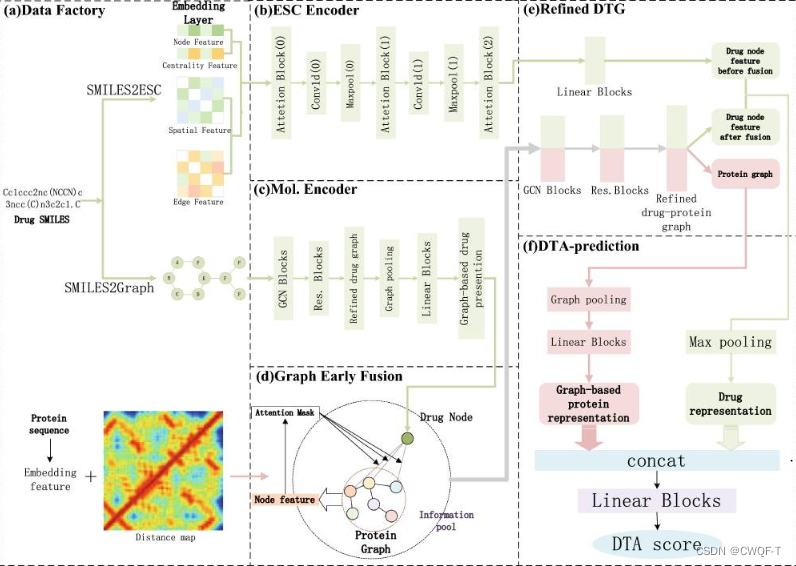
1.药物表示
基于SMILES 和 SDF文件数据来构建分子图(SDF格式的分子数据是使用RDKit工具获得分子的二维结构信息。),包括两大块特征提取器。
2.蛋白质表示
氨基酸序列-->距离图、二维结构、PSSM矩阵、相似性分数矩阵
2.实验
1.数据集
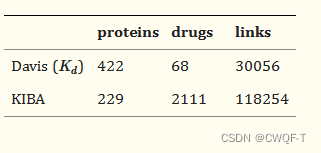
3.结果
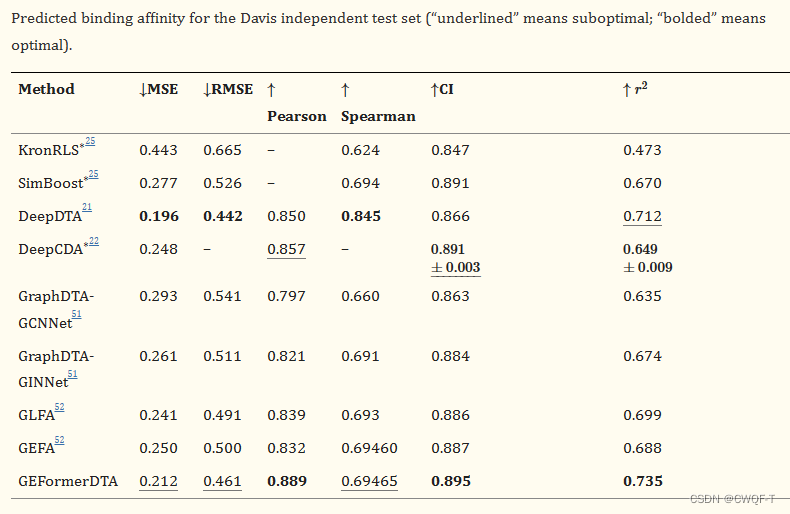
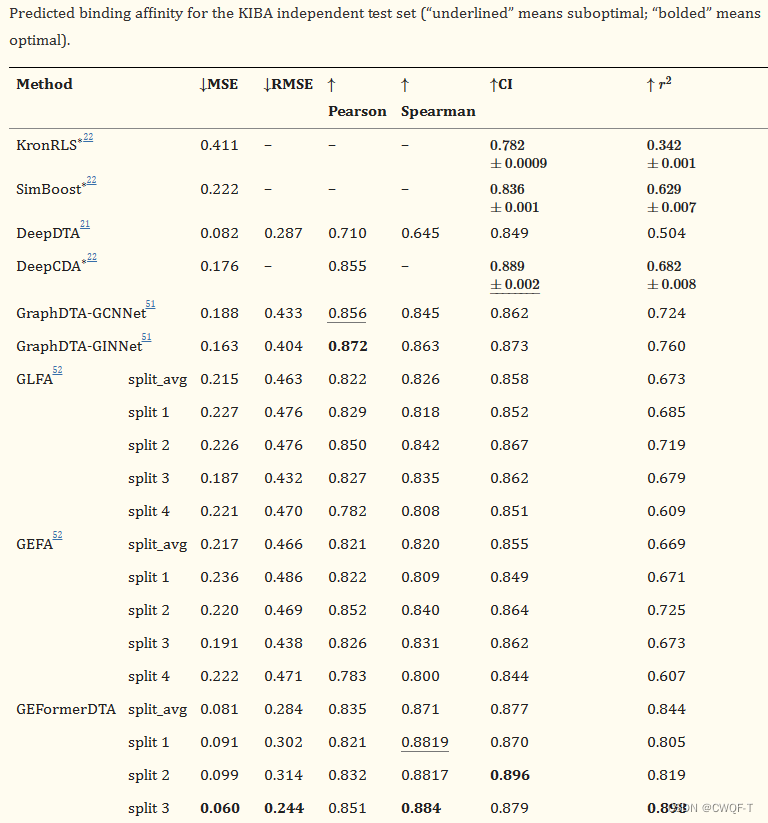
在KIBA数据集的四个子集上用模型进行了实验,所提出的方法在 split3 子集中表现出良好的性能
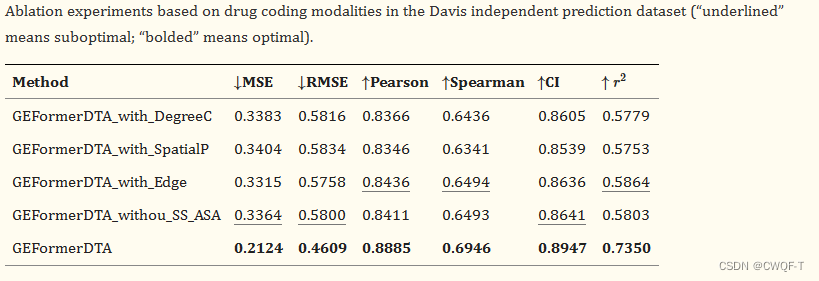
消融实验:没有编码子结构(前三行分别表示没有度心编码、边缘编码和空间编码)的 GEFormerDTA 模型的表现都比同时具有三个编码子结构的模型差。没有蛋白质二级结构和可访问表面积特征编码(第四行)的 GEFormerDTA 模型的表现比具有蛋白质结构特征的模型差。
三.Fusing Sequence and Structural Knowledge by Heterogeneous Models to Accurately and Interpretively Predict Drug–Target Affinity
通过异质模型融合序列和结构知识,准确、解释性地预测药物-靶标亲和力 2023.12 二区
1.模型
通过利用药物 SMILES、靶点和口袋的序列特征及其相应的结构特征来预测药物-靶点亲和力 (DTA)。该模型包括四个基本模块:
(1)数据输入模块负责表示药物、靶点和口袋的序列和结构数据。序列表示的是药物SMILES和氨基酸序列经过标签编码和one-hot编码之后得到的特征向量;结构数据包括药物分子图和靶点图以及结合口袋图(药物图的形成需要.sdf或者.mol文件;靶点图:将靶点结构中氨基酸残基的碳α原子指定为顶点,在欧几里得距离为8 Å的任意两个碳α原子之间建立边而得到靶点图。口袋的结构表示与靶点一致)
(2)在序列学习模块中,采用一维CNN层,根据靶点和口袋的序列数据,从目标和口袋中提取语义特征。
(3)在结构学习模块中,采用3个独立的图卷积网络(GCNs)从药物分子图、靶点和口袋表示中存在的顶点中提取高级特征。
(4)特征融合模块包含两层全连接(FC)网络,通过GCN网络从药物、靶点和口袋中提取的特征进行串联,形成一个全面的256维特征向量。然后将该特征向量输入到 FC 网络中以预测 DTA。
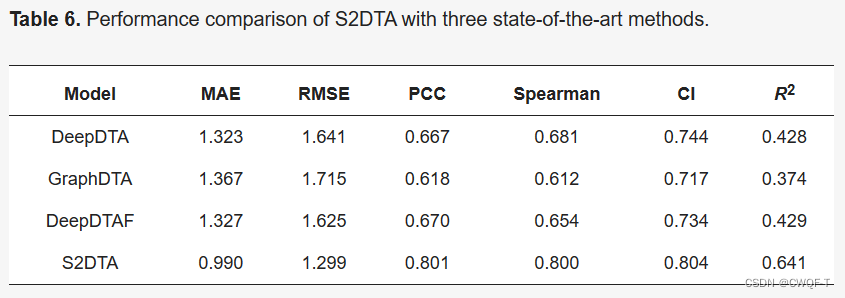
最后,作者还在池化操作之后使用双向注意力机制来提取有关药物-靶标结合亲和力的关键信息,产生了捕获药物和靶标之间相互作用的注意力评分矩阵。通过利用药物和靶点的结构知识为S2DTA的有效性提供可解释性。对于药物,作者计算了靶标中每个原子(不包括氢原子)和所有氨基酸之间的结合数据。随后,进行了数据统计,以确定注意力评分矩阵中的前5、前10和前20个氨基酸是否位于口袋内。统计结果显示,口袋内前5、前10、前20名氨基酸分别占13.7%、12.8%和12.4%。还检查了每个氨基酸与所有原子(不包括氢原子)之间的相互作用。对注意力评分矩阵中的前3、前5和前8原子进行统计分析。这些原子分别占药物原子总数的48.6%、56%和64.8%。所以从双向自注意力中得出的注意力评分矩阵是S2DTA在准确预测药物-靶标结合亲和力方面的有效性的关键解释。
1.药物表示
SMILES和分子图
2.蛋白质表示
氨基酸序列。此外还有结合口袋的序列表示(需要.pdb文件)。
2.实验
1.数据集
PDBbind 数据库(2016 年版)的数据集
3.结果
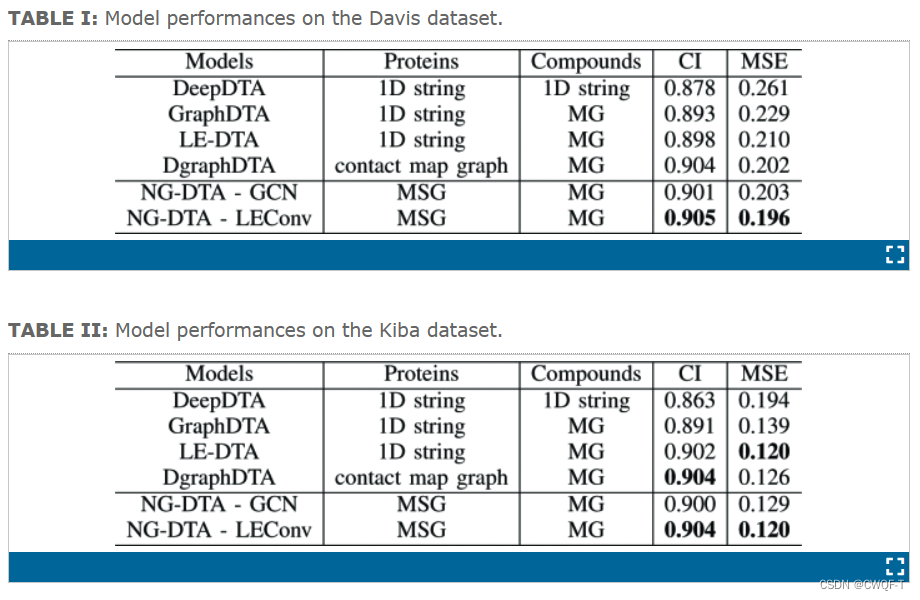
四.NG-DTA: Drug-target affinity prediction with n-gram molecular graphs
使用 n-gram 分子图进行药物靶点亲和力预测 2023.7 2023年第45届IEEE医学与生物学工程学会国际年会
1.模型
利用GNN处理蛋白质的n-gram分子子图和药物的完整分子图。因为蛋白质的分子图对于GNN输入来说太大了,所以没有考虑整个蛋白质分子图,而是将蛋白质转换为n-gram分子子图,这些子图体积小,可以降低复杂性和内存要求。通过使用 n-gram 分子子图作为蛋白质的输入,该模型可以捕获其与有关药物进行相互作用的分子结构。
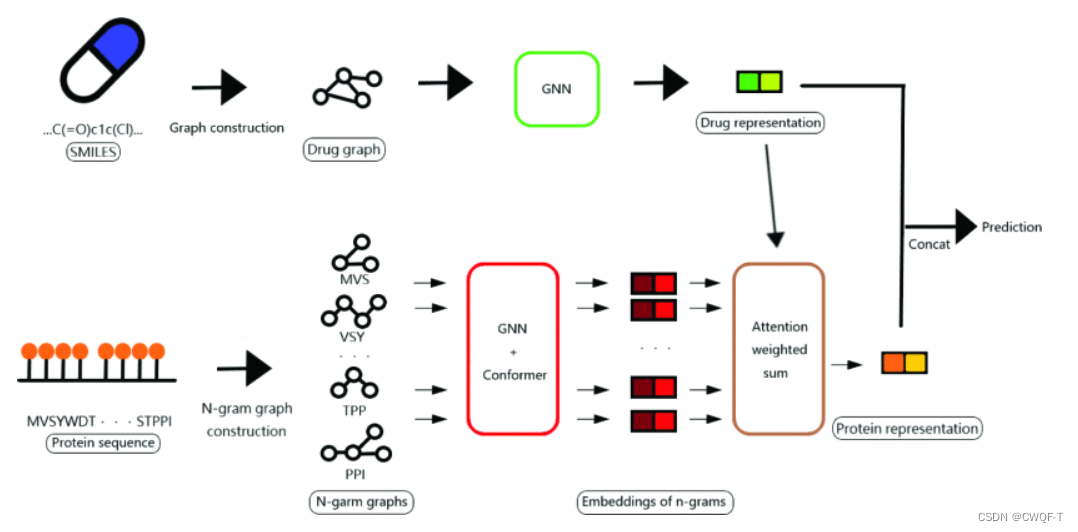 具体来说,使用两个GNN(每个GNN由3个卷积层和一个池化层组成)来获得分子图的图嵌入。GNN中使用了图卷积网络(GCN)和LEConv(可以通过使用自循环和学习局部极值的函数来捕获全局和局部的重要性)。
具体来说,使用两个GNN(每个GNN由3个卷积层和一个池化层组成)来获得分子图的图嵌入。GNN中使用了图卷积网络(GCN)和LEConv(可以通过使用自循环和学习局部极值的函数来捕获全局和局部的重要性)。
蛋白质序列分为重叠的 3-gram。每个 3-gram被转换为一个分子图,之后使用GNN 和 Conformer 对其进行处理。最后,使用注意力加权和来聚合3ram的隐藏表示。注意力加权的计算如下:
[g1, g2, …, gn]是3-gram的图嵌入序列 ,d是药物表示。Watt是可学习的权重,batt是偏置向量,a是第i个3-gram的注意力权重。
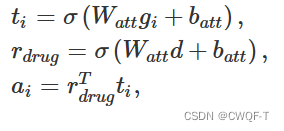
3-gram序列图的注意力加权和表示如下:
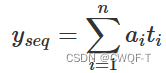
细节图如下:
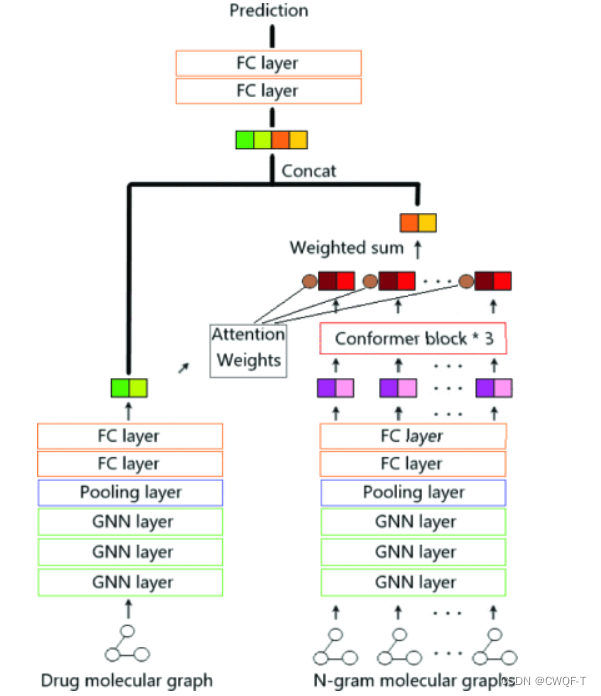
1.药物表示
用RDkit基于SMILES来构建分子图
2.蛋白质表示
设置序列最大长度为1000,长的截断,短的补齐。然后将氨基酸序列转换为重叠的3-gram序列,再使用RDkit将每个3-gram转换为一个分子图,其节点是原子,边是键。即使用蛋白质分子图
2.实验
1.数据集
Davis,Kiba
3.结果
其中,NG-DTA-GCN是使用GCN代替包含LEConv的GNN操作。

















 1593
1593

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









