







在最近半年的工作中,一直在和VCF文件打交道,从一开始的陌生到现在的熟悉,花费了不少功夫,在过程中很多不明白的地方都是到处去收集,反而零零散散,因此我想写一篇博客详细的讲讲vcf文件。
目录
1.什么是VCF文件?
在生物信息学的研究领域,对基因组序列的变异进行精确记录和分析至关重要。VCF(Variant Call Format)文件作为一种标准化的文本格式,为科学家们提供了一种高效的方式来存储和交换基因组变异数据。
VCF格式最初由大名鼎鼎的1000 Genomes Project提出,并随着生物信息学的发展不断完善。目前,VCF格式的维护和扩展工作由全球基因组与健康联盟(Global Alliance for Genomics and Health)的数据工作组负责。
VCF文件用于存储基因组序列变异信息。它能够记录个体基因组中的单核苷酸多态性(SNPs)、插入和缺失(indels)以及其他结构变异。VCF文件的应用范围广泛,包括但不限于进化树分析、群体结构分析、主成分分析(PCA)、全基因组关联研究(GWAS)、二代测序的肿瘤基因组研究等。
2.VCF文件的结构
VCF文件的主要结构如下所示:
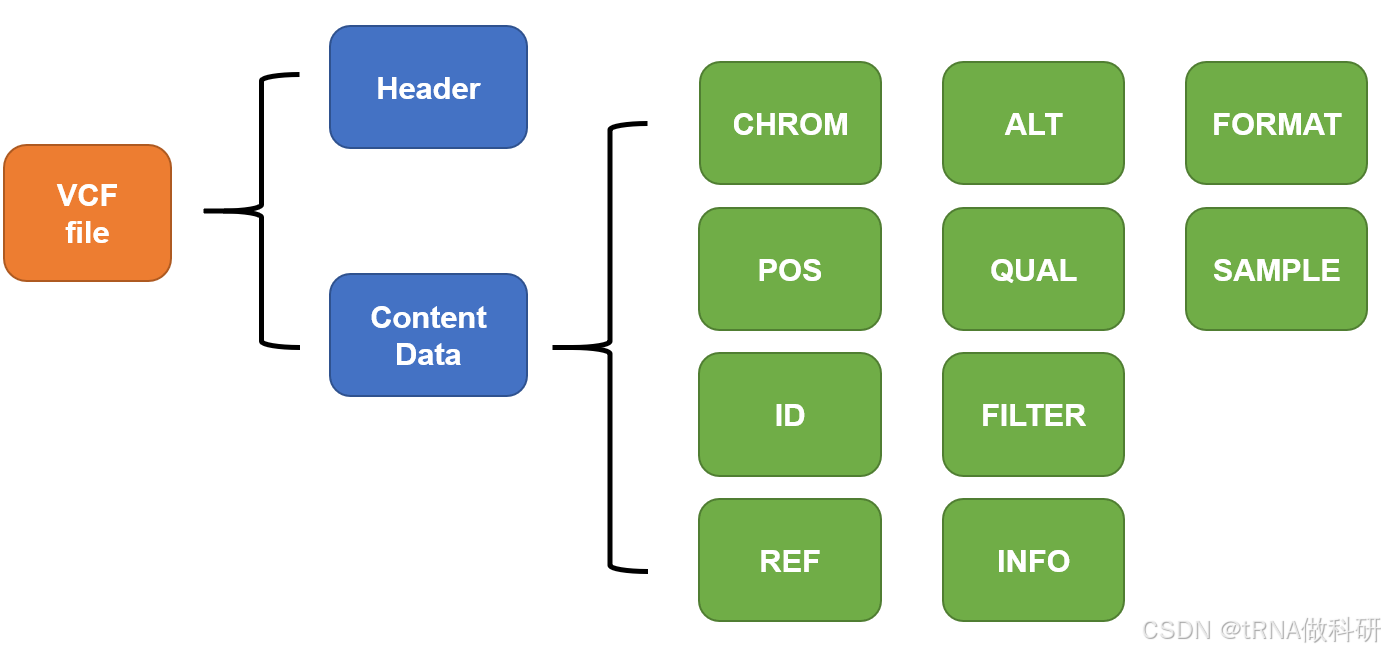
大体由两部分组成:注释部分和主体内容部分。
我们以一个虚构的VCF文件来看:
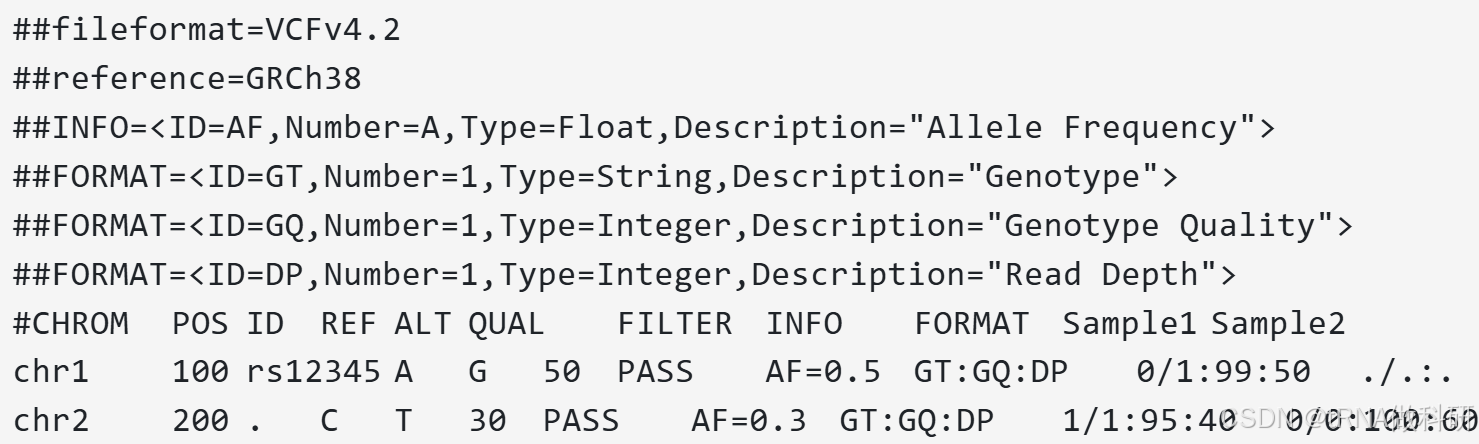
(1)注释部分
注释部分以“##”开头,包含文件格式版本、参考序列、注释信息等元数据。这部分提供了关于VCF文件本身的详细信息,如创建日期、软件版本、参考基因组构建等。
在注释部分中记录了后续内容中每个内容是什么,由什么产生等详细信息,因此是需要阅读的。
例如下:
##fileformat=VCFv4.2
##reference=GRCh38
##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">- 第一行指定了VCF文件的格式版本为VCFv4.2。
- 第二行指定了参考基因组构建为GRCh38。
- 第三行和第四行定义了INFO字段中的AF标签,表示等位基因频率。
- 第五行和第六行定义了FORMAT字段中的GT、GQ和DP标签,分别表示基因型、基因型质量和读取深度。
- 第七行是VCF文件的标题行,列出了所有必需的字段和样本名称。
这些信息有助于我们对vcf进行进一步处理的时候,理解每个数据是什么意思。
(2)标题行
标题行在注释部分后,内容部分前,它定义了文件主体部分每一列的含义。例如:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2(3)内容部分
主体部分记录具体的变异信息,每行代表一个基因位点的变异。每行包含多个字段,这些字段由制表符分隔。例如下:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2
chr1 100 rs12345 A G 50 PASS AF=0.5 GT:GQ:DP 0/1:99:50 ./.:.
chr2 200 . C T 30 PASS AF=0.3 GT:GQ:DP 1/1:95:40 0/0:100:60以下是VCF文件的主要字段:
- CHROM:表示变异位点所在的染色体或contig。
- POS:变异位点在参考基因组中的位置。
- ID:如果变异在数据库中有记录,则给出相应的标识符,如dbSNP中的rs编号;否则用“.”表示。
- REF:参考基因组中的碱基。
- ALT:变异基因组中的碱基。
- QUAL:Phred格式的质量值,表示变异位点的可靠性。
- FILTER:表示变异位点是否通过了质量控制过滤。
- INFO:包含多个以分号分隔的键值对,提供关于变异的额外信息,如等位基因频率(AF)、深度(DP)等。
- FORMAT:定义了后续样本列中数据的格式,常见的有GT(基因型)、GQ(基因型质量)、DP(测序深度)等。
- 样本基因型信息:每个样本的基因型信息,按照FORMAT字段指定的格式排列。
了解了这些就基本了解了VCF文件的格式,我们进一步仔细学习一下INFO和FORMAT的内容
3.INFO信息
INFO字段是VCF文件中的一个重要组成部分,它用于存储与变异位点相关的附加信息。这些信息通常以键值对的形式存在,每个键值对由等号(=)连接,而不同的键值对之间则由分号(;)分隔。INFO字段可以包含多种类型的统计数据和注释信息,这些信息对于理解变异位点的性质和影响至关重要。例如:
AF=0.2;AC=4;DP=50;VT=SNP;IMPACT=Moderate常见的字段包括:
- 等位基因频率(AF):表示变异位点的次生等位基因在样本集合中的频率。
- 等位基因计数(AC):表示变异位点的次生等位基因在样本集合中出现的次数。
- 基因型频率(GF):表示不同基因型在样本集合中的分布情况。
- 深度(DP):表示测序数据在变异位点的覆盖深度。
- 质量得分(QUAL):虽然通常在VCF文件的独立字段中给出,但有时也会在INFO字段中提供更详细的得分信息。
- 过滤状态(FILTER):虽然也有独立的FILTER字段,但INFO字段可能会包含更多关于过滤原因的细节。
- 变异类型(VT):表示变异是SNP、indel还是其他类型。
- 影响(IMPACT):表示变异对基因或蛋白质功能的潜在影响程度。
- 来源(SRC):表示变异是如何被检测到的,例如通过哪种测序技术或分析方法。
- 注释(ANN):提供关于变异位点的详细生物学注释,如受影响的基因、蛋白改变等。
以我们的例子来看:
AF=0.2 表示等位基因频率为0.2,AC=4 表示等位基因计数为4,DP=50 表示测序深度为50,VT=SNP 表示这是一个单核苷酸多态性,而 IMPACT=Moderate 表示这个变异对基因或蛋白质功能的影响程度为中等。
INFO字段提供了变异位点的详细背景信息,这对于后续的分析和解释至关重要。例如,在进行全基因组关联研究(GWAS)时,等位基因频率和等位基因计数可以帮助识别与疾病相关的变异。在进行基因组注释时,注释信息可以帮助了解变异对基因表达和蛋白质功能的潜在影响。
4.FORMAT字段
FORMAT字段在VCF文件中用于指定样本数据的格式。它定义了后续样本列中数据的排列顺序和类型,确保了样本数据的一致性和可读性。FORMAT字段通常包含一系列的标签,每个标签都对应一种特定的数据类型,如基因型(GT)、基因型质量(GQ)、测序深度(DP)等。
其实就是INFO主要是针对位点,而FORMAT代表的是每个样本个性化的信息!
FORMAT字段可能包含以下标签:
- GT(Genotype):表示样本在该变异位点的基因型。对于二倍体生物,基因型通常表示为两个数字的组合,如“0/1”表示杂合子,其中“0”代表参考等位基因,“1”代表变异等位基因。
- GQ(Genotype Quality):表示基因型质量得分,反映了对基因型推断的置信度。得分越高,表示基因型推断的准确性越高。
- DP(Depth):表示测序深度,即在该变异位点的测序覆盖度。DP值越高,表示该位点的测序数据越丰富,推断结果越可靠。
- AD(Allelic Depth):表示每个等位基因的测序深度。对于二倍体生物,AD通常包含两个数字,分别表示参考等位基因和变异等位基因的读取次数。
- PL(Phred-scaled Likelihoods):表示基于Phred尺度的似然比,用于评估不同基因型的可能性。PL值越低,表示对应的基因型可能性越高。
我们举个例子说明:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2
chr1 10001 rs12345 A C 50 PASS AF=0.2;DP=30 GT:GQ:DP 0/1:99:15 ./.:.
chr1 10002 . T G 30 FILTERED AF=0.5;DP=20 GT:GQ:DP 1/1:95:10 0/0:100:25 在这个例子中,FORMAT字段为“GT:GQ:DP”,这意味着每个样本的数据都将按照“基因型:基因型质量:测序深度”的顺序排列。
对于第一个变异位点(chr1:10001),Sample1的数据显示为“0/1:99:15”,这表示:
GT(基因型):Sample1在这个位点的基因型为杂合子(0/1),其中“0”代表参考等位基因“A”,“1”代表变异等位基因“C”。
GQ(基因型质量):Sample1的基因型质量得分为99,这是一个很高的得分,表明对该基因型的推断非常有信心。
DP(测序深度):Sample1在这个位点的测序深度为15,这意味着有15条读段覆盖了这个位点。
对于第二个变异位点(chr1:10002),Sample1的数据显示为“1/1:95:10”,这表示:
GT(基因型):Sample1在这个位点的基因型为纯合子(1/1),其中两个“1”都代表变异等位基因“G”。
GQ(基因型质量):Sample1的基因型质量得分为95,这也是一个很高的得分,表明对该基因型的推断非常有信心。
DP(测序深度):Sample1在这个位点的测序深度为10,这意味着有10条读段覆盖了这个位点。
同时,Sample2在第一个变异位点没有数据(显示为“./.:.”),这可能意味着样本没有被成功测序或者数据质量不足以进行推断。而在第二个变异位点,Sample2的数据显示为“0/0:100:25”,这表示Sample2在这个位点的基因型为纯合子(0/0),基因型质量得分为100,测序深度为25。
5.针对GATK的VCF文件理解
对于最新的GATK call mutation的教程后面会更新,不过这里我简要叙述一下mutect2及后续mutect2质控后得到的VCF文件的一些基本信息:
其INFO和FORMAT例如下:
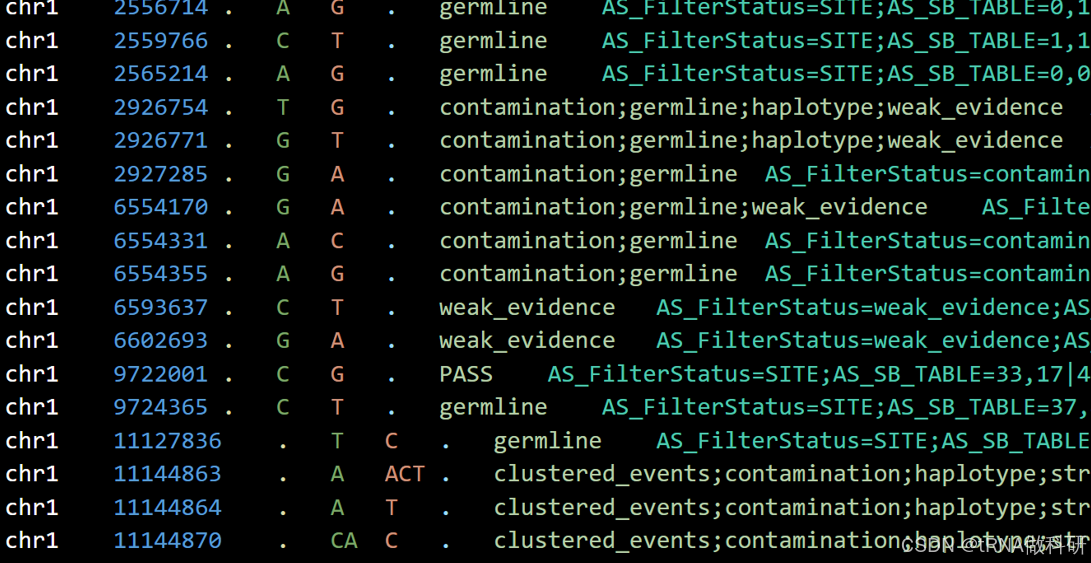
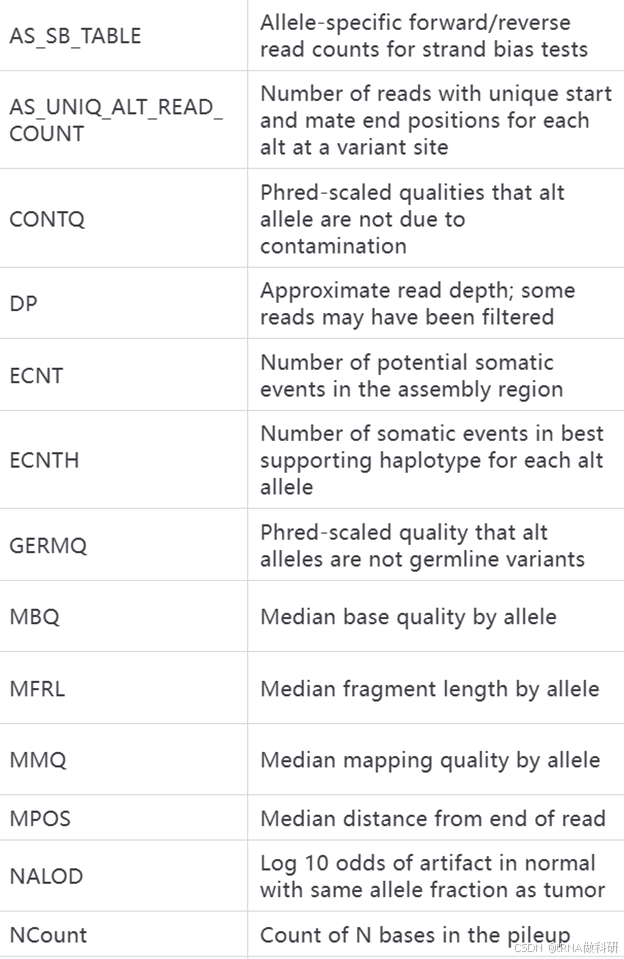
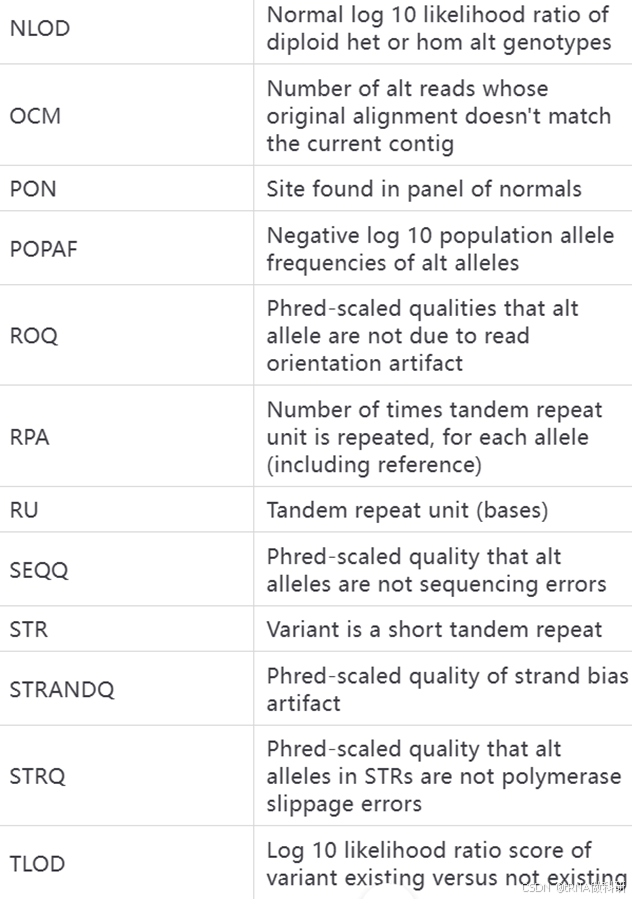
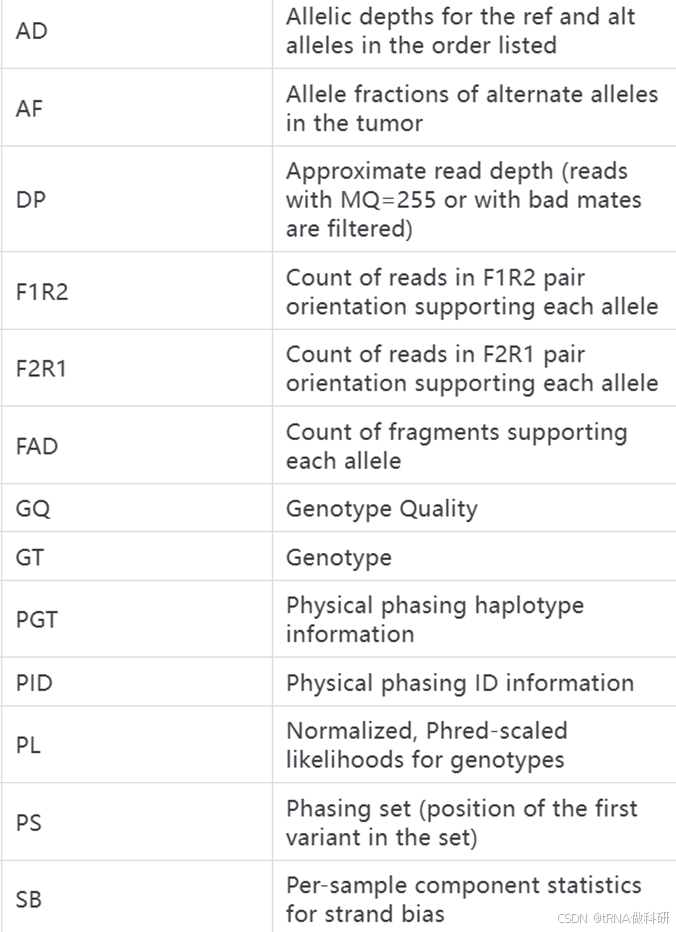
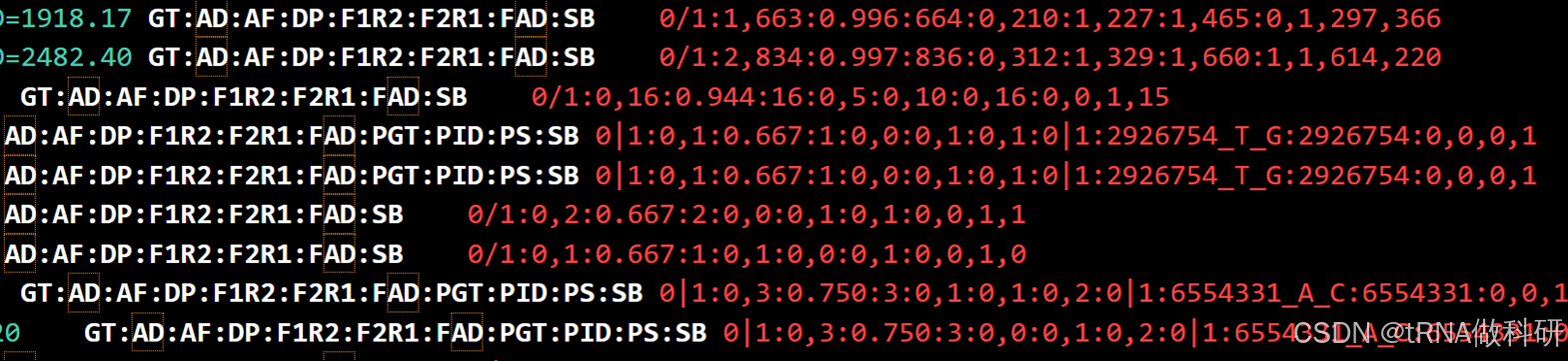 这些INFO和FORMAT代表的含义我总结了表格如下:
这些INFO和FORMAT代表的含义我总结了表格如下:
INFO
FORMAT
6.GATK对于mutation的FILTER
GATK对于VCF的质控后,会将结果存放在FILTER列中,方便后续的查找,我总结了几种常见的:


后续的更详细的内容会出针对GATK筛选流程的博客!如果希望我早点出可以评论区和我说!
7.一些操作VCF的软件-VCFTOOLS
我们以VCFtools为例子讲述一些VCF文件的基本操作:
#首先安装vcftools
conda install vcftools
示例1:过滤VCF文件中的SNPs
vcftools --vcf input.vcf --remove-indels --recode --out filteredsnps 这个命令会从input.vcf文件中移除所有的插入和缺失(indels),只保留SNPs,并将结果输出到filteredsnps.vcf文件中。
示例2:计算群体的核苷酸多样性
vcftools --vcf input.vcf --diversity --out diversitystats这个命令会计算input.vcf文件中群体的核苷酸多样性,并将统计结果输出到diversitystats文件中。
示例3:比较两个VCF文件的差异 bash
vcftools --vcf file1.vcf --diff file2.vcf --diff-site --out Diff.site这个命令会比较file1.vcf和file2.vcf两个文件中的变异位点,并将差异输出到Diff.site文件中。
示例4:提取特定区域的VCF数据
vcftools --vcf input.vcf --bed regions.bed --out extractedvariants这个命令会从input.vcf文件中提取位于regions.bed文件指定区域内的变异,并将结果输出到extractedvariants.vcf文件中。
示例5:提取特定样本的VCF数据
vcftools --vcf input.vcf --ids samples.txt --out specificsamples 这个命令会从input.vcf文件中提取samples.txt文件列出的样本的变异数据,并将结果输出到specificsamples.vcf文件中。
示例6:计算缺失率
vcftools --vcf input.vcf --missing-indv --out missingness这个命令会计算input.vcf文件中每个样本的缺失率,并将结果输出到missingness文件中。
上述命令中的--out选项后面跟的是输出文件的前缀,实际的输出文件名会在这个前缀基础上添加相应的扩展名。
如果还有什么问题可以评论区与我讨论!



















 2317
2317

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











