







https://smorabit.github.io/hdWGCNA/index.html
https://mp.weixin.qq.com/s/OBvS0I7IUuwcChGoaJVgtQ
https://smorabit.github.io/hdWGCNA/articles/basic_tutorial.html#co-expression-network-analysis
load("D:/ARDS_scripts_1012/ARDS/Step2_harmony_f200_R3/0805/cluster_merge/sepsis_cluster_merge.rds")## 改路径
table(All.merge$stim)
table(Idents(All.merge),All.merge$stim)
1创建对象,选择基因
length(VariableFeatures(All.merge))
seurat_obj <- SetupForWGCNA(
All.merge,
gene_select = "fraction", # the gene selection approach
fraction = 0.05, # use genes that are expressed in a certain fraction of cells for in the whole dataset or in each group of cells, specified by group.by fraction of cells that a gene needs to be expressed in order to be included
#variable=
wgcna_name = "tutorial" # the name of the hdWGCNA experiment
)
length(seurat_obj@misc t u t o r i a l tutorial tutorialwgcna_genes) #[1] 5735
table(seurat_obj$cell.type)
table(seurat_obj$stim)
seurat_obj$Sample=seurat_obj$stim
seurat_obj$cell_type=seurat_obj$cell.type

2# construct metacells in each group
seurat_obj <- MetacellsByGroups(
seurat_obj = seurat_obj,
group.by = c("cell_type", "Sample"), # specify the columns in seurat_obj@meta.data to group by
k = 20, # nearest-neighbors parameter
reduction = "harmony", # 降维方法
slot='counts',
max_shared = 10, # maximum number of shared cells between two metacells
ident.group = 'cell_type' # set the Idents of the metacell seurat object
)
3#标准化:NormalizeMetacells
#提取metacell:GetMetacellObject
# normalize metacell expression matrix:
seurat_obj <- NormalizeMetacells(seurat_obj)
# get the metacell object from the hdWGCNA experiment
metacell_obj <- GetMetacellObject(seurat_obj)
metacell_obj
seurat_obj
table(seurat_obj$cell_type,seurat_obj$Sample)
table(metacell_obj$cell_type,metacell_obj$Sample)


3.1#可选,是否处理metacell 进行可视化
if(F){
seurat_obj <- NormalizeMetacells(seurat_obj)
seurat_obj <- ScaleMetacells(seurat_obj, features=VariableFeatures(seurat_obj))
seurat_obj <- RunPCAMetacells(seurat_obj, features=VariableFeatures(seurat_obj))
seurat_obj <- RunHarmonyMetacells(seurat_obj, group.by.vars='Sample')
seurat_obj <- RunUMAPMetacells(seurat_obj, reduction='harmony', dims=1:15)
p1 <- DimPlotMetacells(seurat_obj, group.by='cell_type') + umap_theme() + ggtitle("Cell Type")
p2 <- DimPlotMetacells(seurat_obj, group.by='Sample') + umap_theme() + ggtitle("Sample")
p1 | p2
}
4#共表达网络分析
table(seurat_obj$cell_type)
table(metacell_obj$cell_type)

4.1#提取感兴趣的细胞亚群 如果是多个亚群
group_name = c("INH", "EX")
seurat_obj <- SetDatExpr(
seurat_obj,
group_name = c("Monocyte","Macrophage","Dendritic cell","Neutrophil",
"T cell","Fibroblast","Endothelial cell",
"Epithelial cell","B cell",
"Mesenchymal progenitor cell"), # the name of the group of interest in the group.by column
group.by='cell_type', # the metadata column containing the cell type info. This same column should have also been used in MetacellsByGroups
assay = 'RNA', # using RNA assay
use_metacells = TRUE, # use the metacells (TRUE) or the full expression matrix (FALSE)
slot = 'data' # using normalized data
)
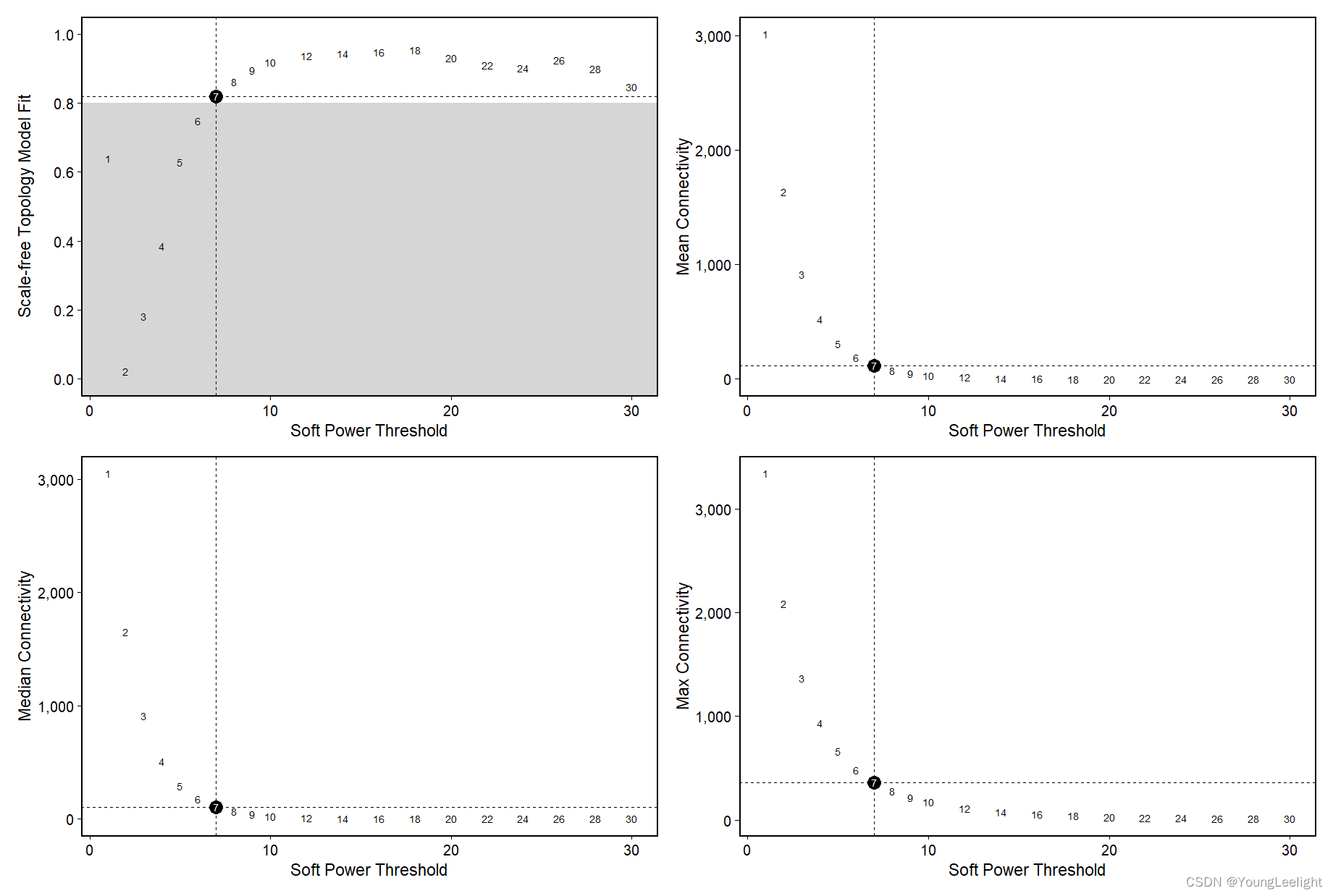
4.2#选取软阈值 软阈值可以自动函数选择,也可以人工指定selected_power = NULL进行绘图
Test different soft powers:
seurat_obj <- TestSoftPowers(
seurat_obj,
setDatExpr=FALSE,
powers = c(seq(1, 10, by = 1), seq(12, 30, by = 2))) # 选取soft powers(默认)
# networkType = 'signed' # 网络类型 "unsigned" or "signed hybrid"
# plot the results:
plot_list <- PlotSoftPowers(seurat_obj,
point_size = 5,
text_size = 3)
# assemble with patchwork
wrap_plots(plot_list, ncol=2)
# plot the results:
plot_list <- PlotSoftPowers(seurat_obj)
# assemble with patchwork
wrap_plots(plot_list, ncol=2)
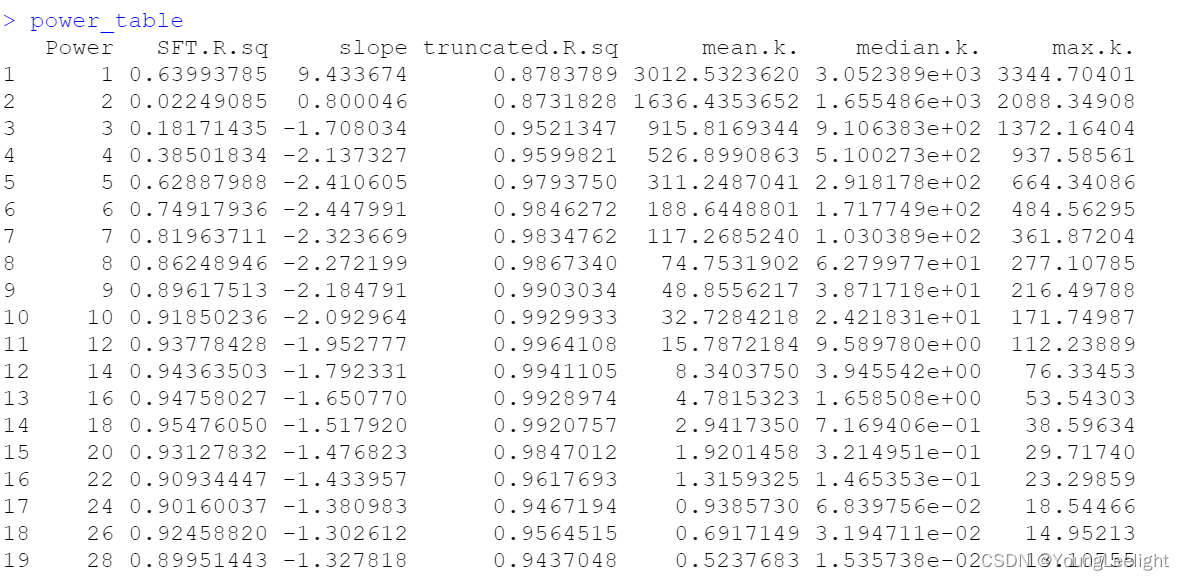
power_table <- GetPowerTable(seurat_obj)
head(power_table)
4.3#拓扑重叠矩阵(TOM) Construct co-expression network
seurat_obj <- ConstructNetwork(
seurat_obj,
soft_power=7, # 因为上面一张图看上去7比较好
setDatExpr=FALSE,
corType = "pearson",
networkType = "signed",
TOMType = "signed",
detectCutHeight = 0.995,
minModuleSize = 50,
mergeCutHeight = 0.2,
tom_outdir = "TOM", # 输出文件夹
tom_name = 'many_celltypes' # name of the topoligical overlap matrix written to disk
)
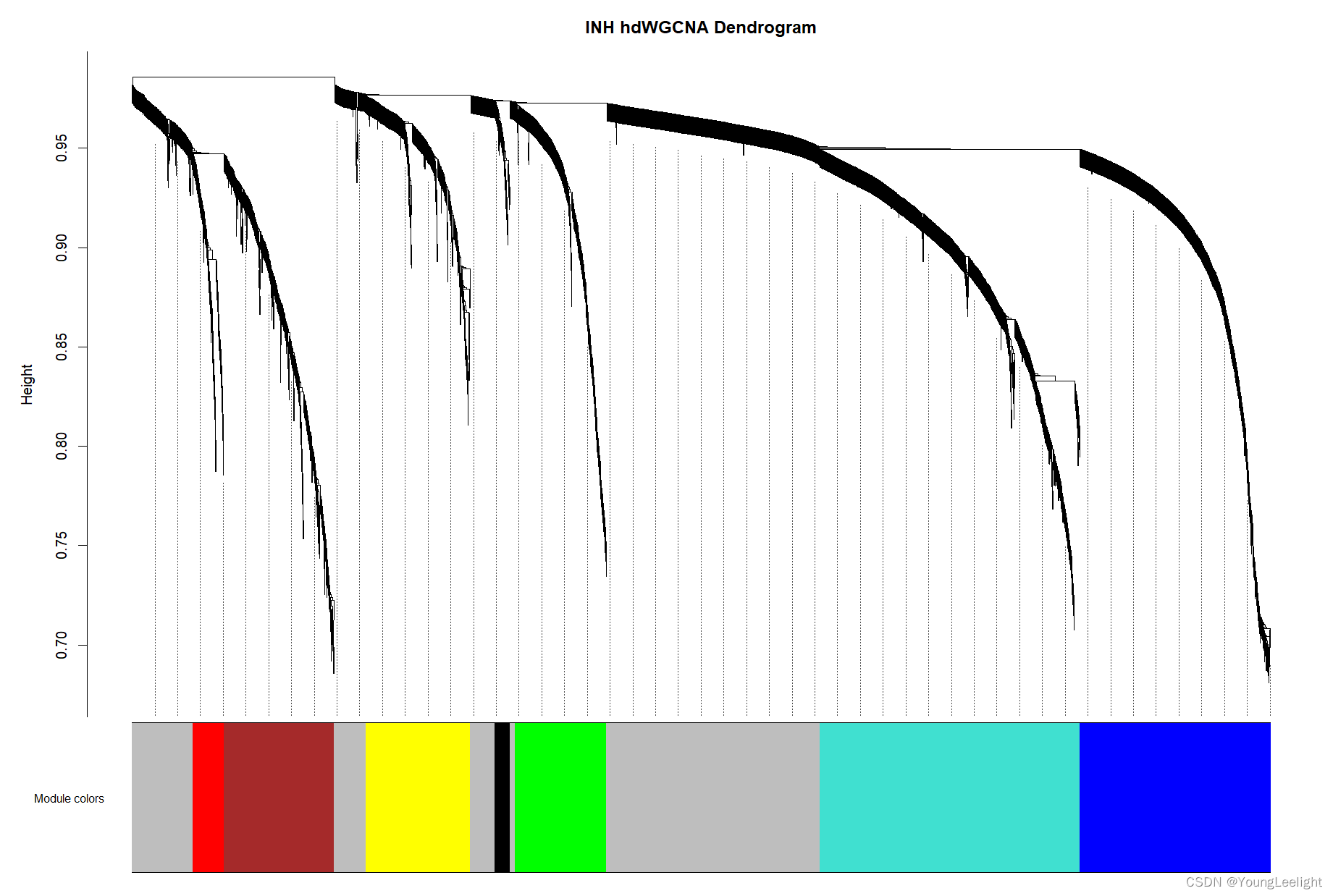
PlotDendrogram(seurat_obj, main='INH hdWGCNA Dendrogram')
4.3.1 可选
#TOM <- GetTOM(seurat_obj)
4.4#计算ME值
DefaultAssay(seurat_obj)
length(VariableFeatures(seurat_obj))
# need to run ScaleData first or else harmony throws an error:
seurat_obj <- ScaleData(seurat_obj, features=VariableFeatures(seurat_obj))
# compute all MEs in the full single-cell dataset
seurat_obj <- ModuleEigengenes(
seurat_obj,
scale.model.use = "linear", # choices are "linear", "poisson", or "negbinom"
assay = NULL, # 默认:DefaultAssay(seurat_obj)
pc_dim = 1,
group.by.vars="Sample" # 根据样本去批次化 harmonize
)
# harmonized module eigengenes:
hMEs <- GetMEs(seurat_obj)
# module eigengenes:
MEs <- GetMEs(seurat_obj, harmonized=FALSE)
colnames(seurat_obj@misc[["tutorial"]][["hMEs"]])
seurat_obj@misc[["tutorial"]][["MEs"]][1:6,1:6]
seurat_obj@misc[["tutorial"]][["hMEs"]][1:6,1:6]
这里的列名是被后面重新命名的 本来是颜色的标签

4.5#模块与性状间的相关性
#其实本质上还是设置成二分类变量或者数字,这样得到的结果就具有生物学意义
if(T){
# convert sex to factor
seurat_obj$msex <- as.factor(seurat_obj$stim)
# convert age_death to numeric
seurat_obj$age_death <- as.numeric(seurat_obj$seurat_clusters)
# list of traits to correlate
head(seurat_obj@meta.data)
cur_traits <- c( 'msex', 'age_death',
'nCount_RNA', 'nFeature_RNA', 'percent.mt')
str(seurat_obj@meta.data[,cur_traits])
# 使用去批次化后的hME
seurat_obj <- ModuleTraitCorrelation(
seurat_obj,
traits = cur_traits,
features = "hMEs", # Valid choices are hMEs, MEs, or scores
cor_method = "pearson", # Valid choices are pearson, spearman, kendall.
group.by='cell_type'
)
# get the mt-correlation results
mt_cor <- GetModuleTraitCorrelation(seurat_obj)
names(mt_cor)
“cor” “pval” “fdr”
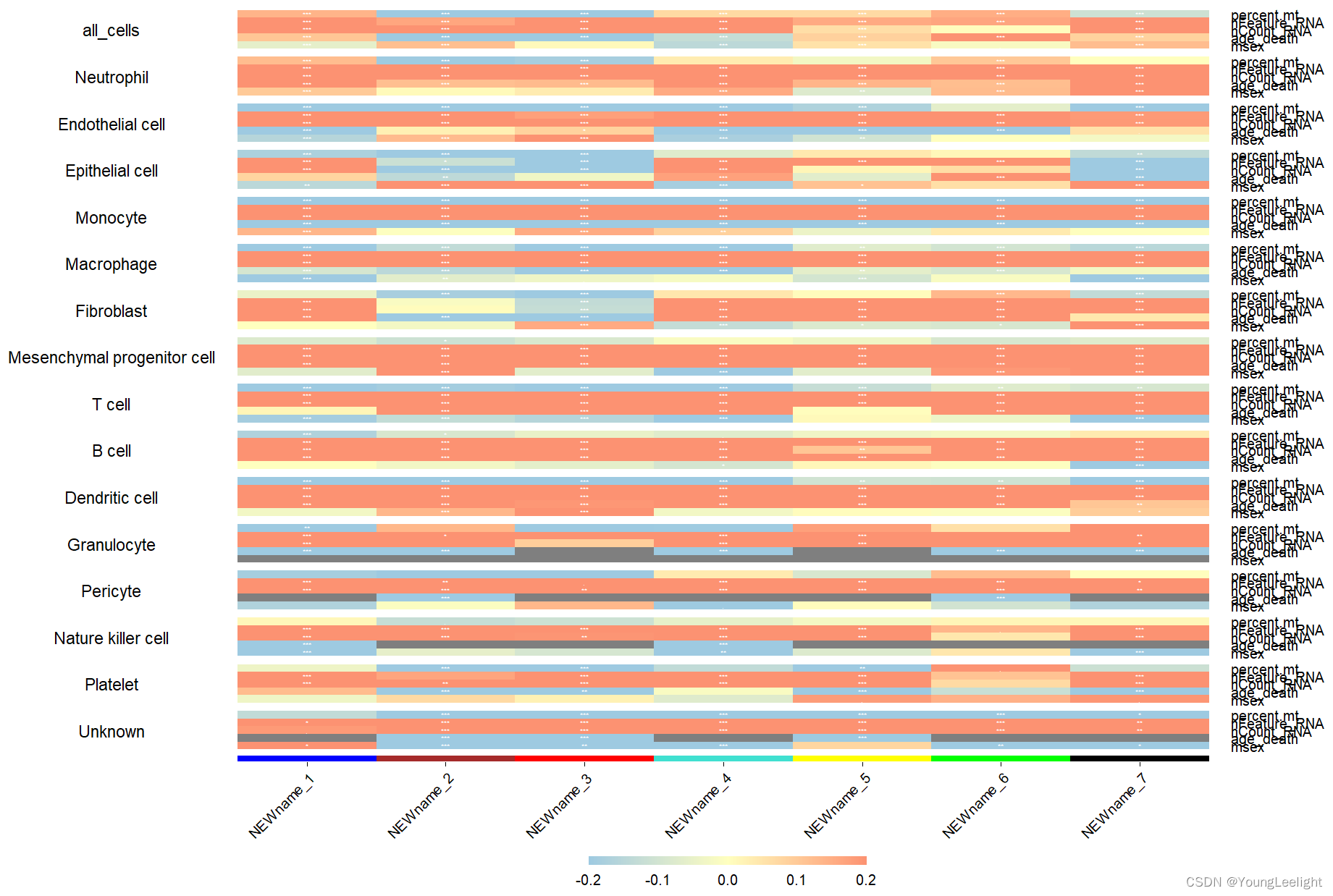
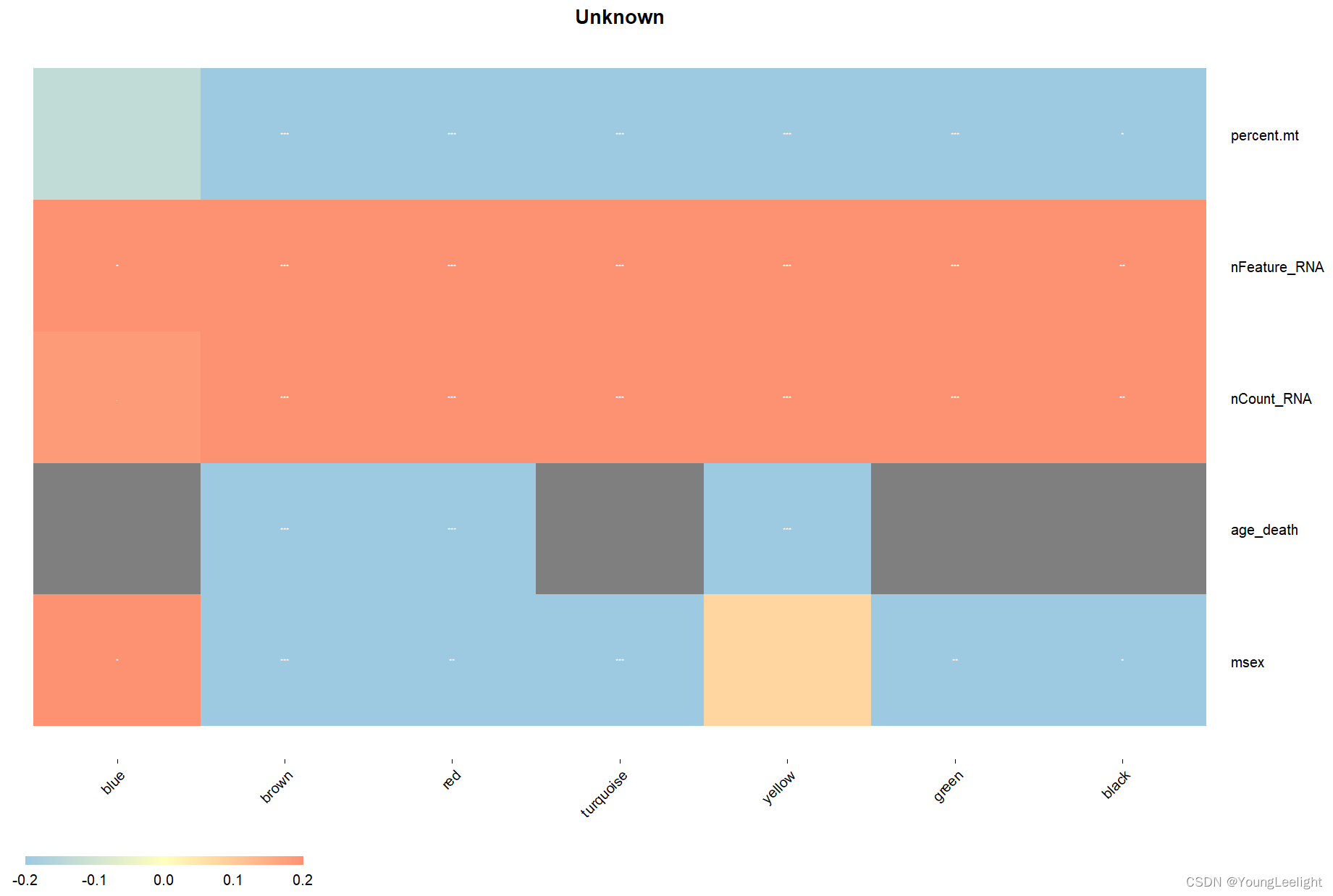
PlotModuleTraitCorrelation(
seurat_obj,
label = 'fdr', # add p-val label in each cell of the heatmap
label_symbol = 'stars', # labels as 'stars' or as 'numeric'
text_size = 2,
text_digits = 2,
text_color = 'white',
high_color = '#fc9272',
mid_color = '#ffffbf',
low_color = '#9ecae1',
plot_max = 0.2,
combine=T # 合并结果
)
PlotModuleTraitCorrelation(
seurat_obj,
label = 'fdr', # add p-val label in each cell of the heatmap
label_symbol = 'stars', # labels as 'stars' or as 'numeric'
text_size = 2,
text_digits = 2,
text_color = 'white',
high_color = '#fc9272',
mid_color = '#ffffbf',
low_color = '#9ecae1',
plot_max = 0.2,
combine=F # 合并结果
)
}
4.6 #hubgenes和功能评分 compute eigengene-based connectivity (kME):
# compute eigengene-based connectivity (kME):
seurat_obj <- ModuleConnectivity(
seurat_obj,
group.by = 'cell_type',
corFnc = "bicor", # to obtain Pearson correlation
corOptions = "use='p'", # to obtain Pearson correlation
harmonized = TRUE,
assay = NULL,
slot = "data", # default to normalized 'data' slot
group_name = c("Monocyte","Macrophage","Dendritic cell","Neutrophil",
"T cell","Fibroblast","Endothelial cell",
"Epithelial cell","B cell",
"Mesenchymal progenitor cell") # 感兴趣的细胞群
)
rename the modules
# 改名后后模块赋予新的名称,以new_name为基础,后续附加个数字
seurat_obj <- ResetModuleNames( #https://smorabit.github.io/hdWGCNA/articles/customization.html
seurat_obj,
new_name = "NEWname_" # the base name for the new modules
)
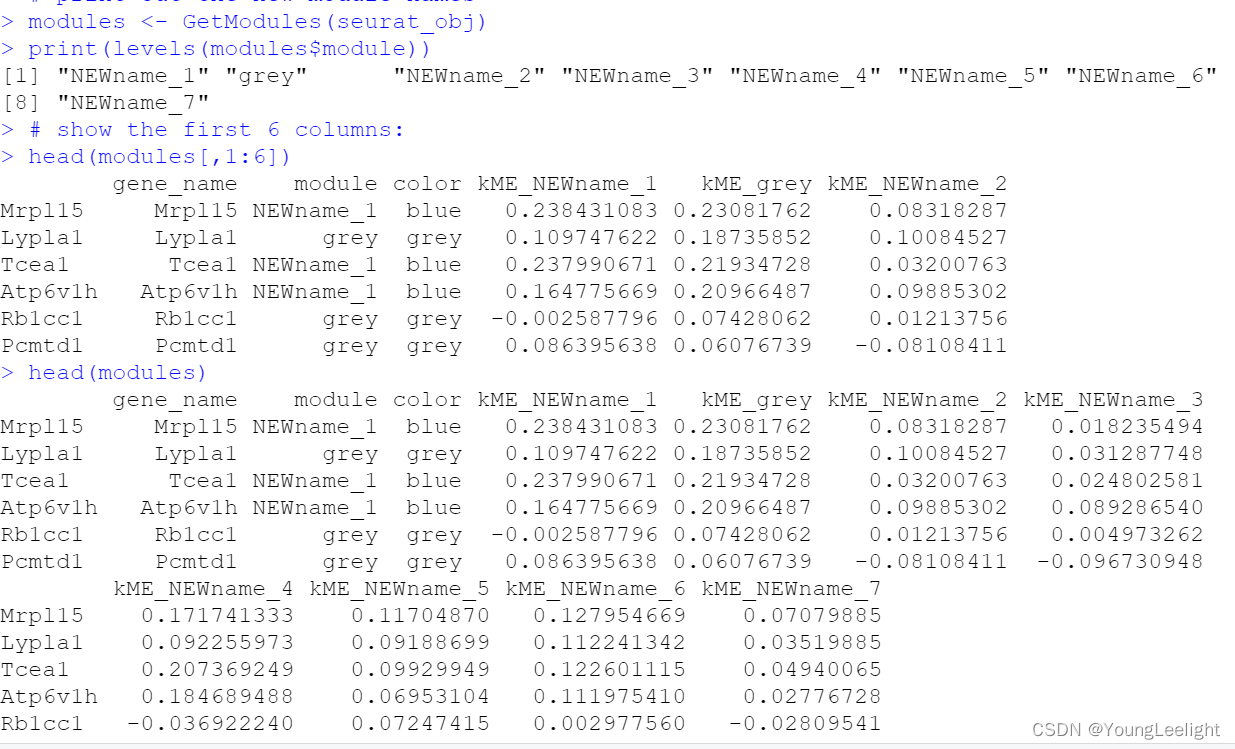
print out the new module names
modules <- GetModules(seurat_obj)
print(levels(modules$module))
# show the first 6 columns:
head(modules[,1:6])
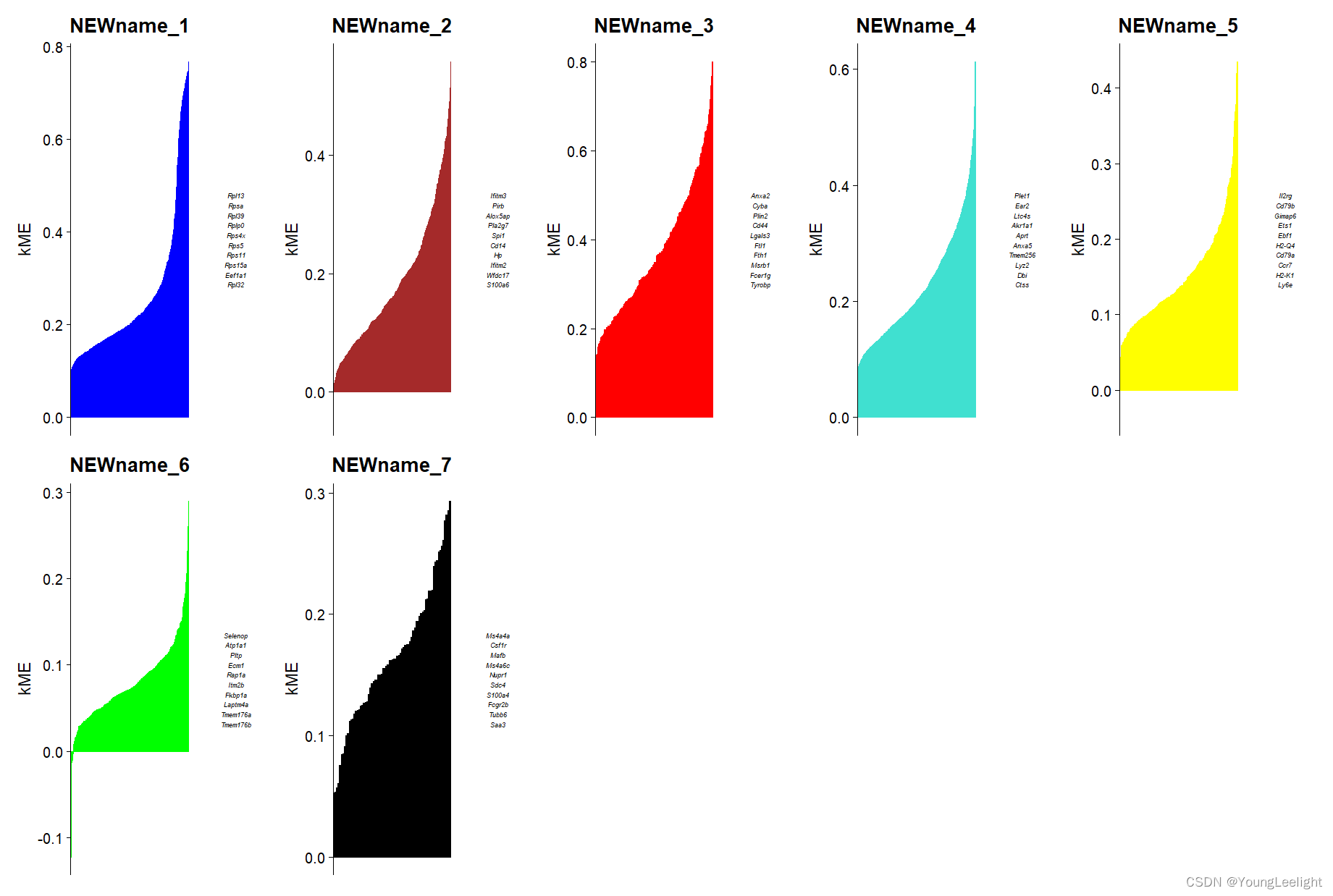
plot genes ranked by kME for each module
p <- PlotKMEs(seurat_obj,
ncol=5,
n_hubs = 10, # number of hub genes to display
text_size = 2,
plot_widths = c(3, 2) # the relative width between the kME rank plot and the hub gene text
)
p
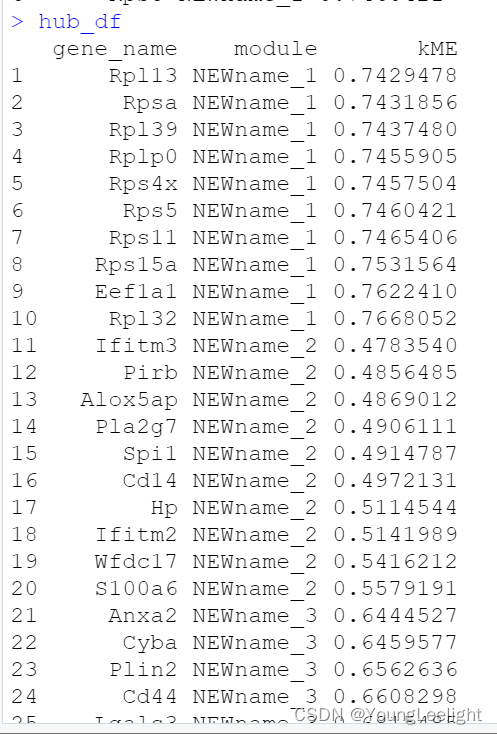
get hub genes
hub_df <- GetHubGenes(seurat_obj, n_hubs = 10)
head(hub_df)
dir.create(“./data”)
saveRDS(seurat_obj, file=‘data/hdWGCNA_object.rds’)
4.6.2 #基因功能评分
# compute gene scoring for the top 25 hub genes by kME for each module
# with Seurat method
seurat_obj <- ModuleExprScore(
seurat_obj,
n_genes = 25,
method='Seurat'
)
compute gene scoring for the top 25 hub genes by kME for each module
with UCell method
library(UCell)
seurat_obj <- ModuleExprScore(
seurat_obj,
n_genes = 25,
method='UCell'
)
4.7#可视化
make a featureplot of hMEs for each module
#其实features可以展示四种分数(hMEs, MEs, scores, or average)
plot_list <-ModuleFeaturePlot(
seurat_obj,
reduction = "umap",
features = "hMEs",
#features='scores', # plot the hub gene scores
order_points = TRUE, # order so the points with highest hMEs are on top
restrict_range = TRUE,
point_size = 0.5,
alpha = 1,
label_legend = FALSE,
raster_dpi = 500,
raster_scale = 1,
plot_ratio = 1,
title = TRUE
)
stitch together with patchwork
wrap_plots(plot_list, ncol=6)
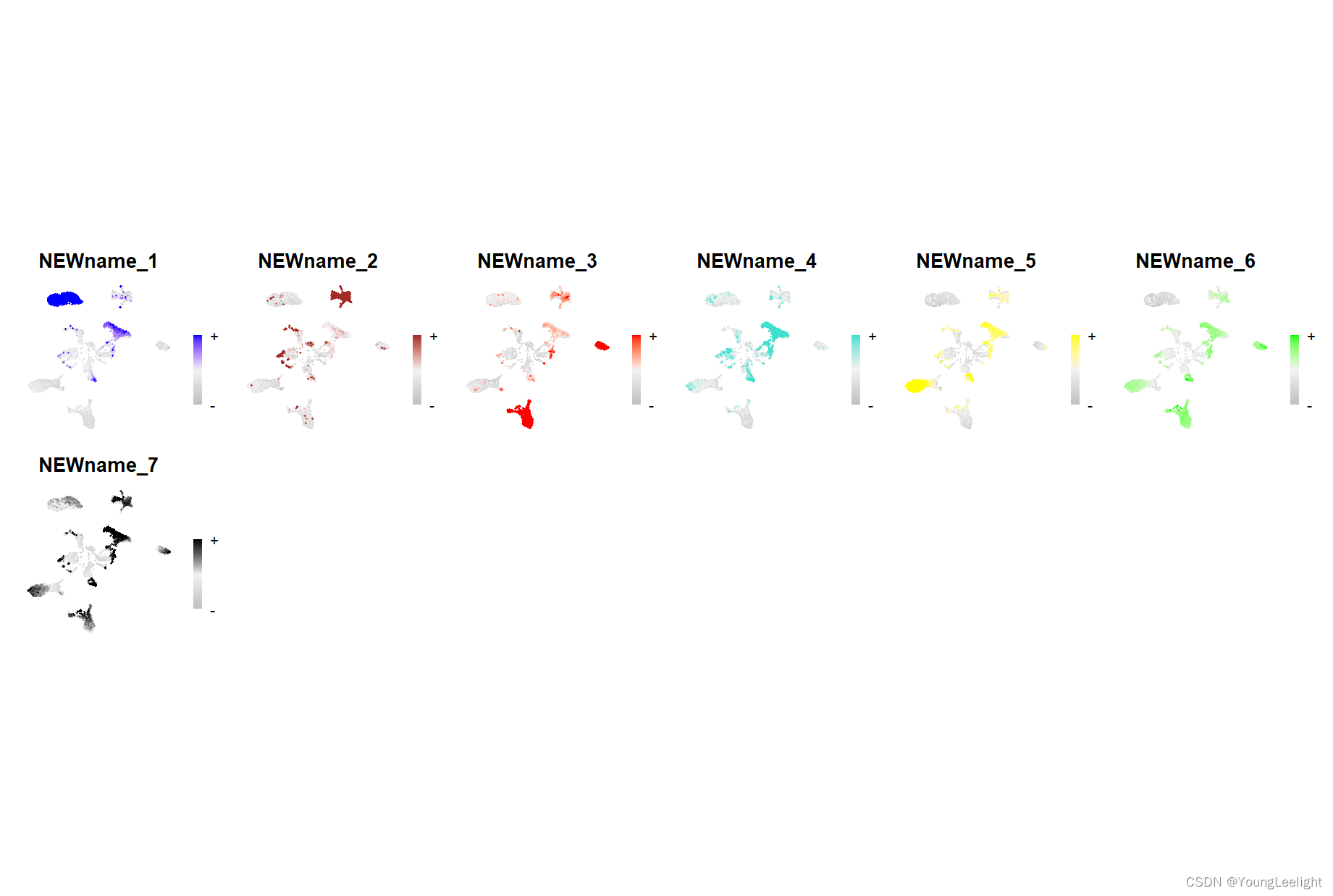
# make a featureplot of hub scores for each module
plot_list <- ModuleFeaturePlot(
seurat_obj,
features='scores', # plot the hub gene scores
order='shuffle', # order so cells are shuffled
ucell = TRUE # depending on Seurat vs UCell for gene scoring
)
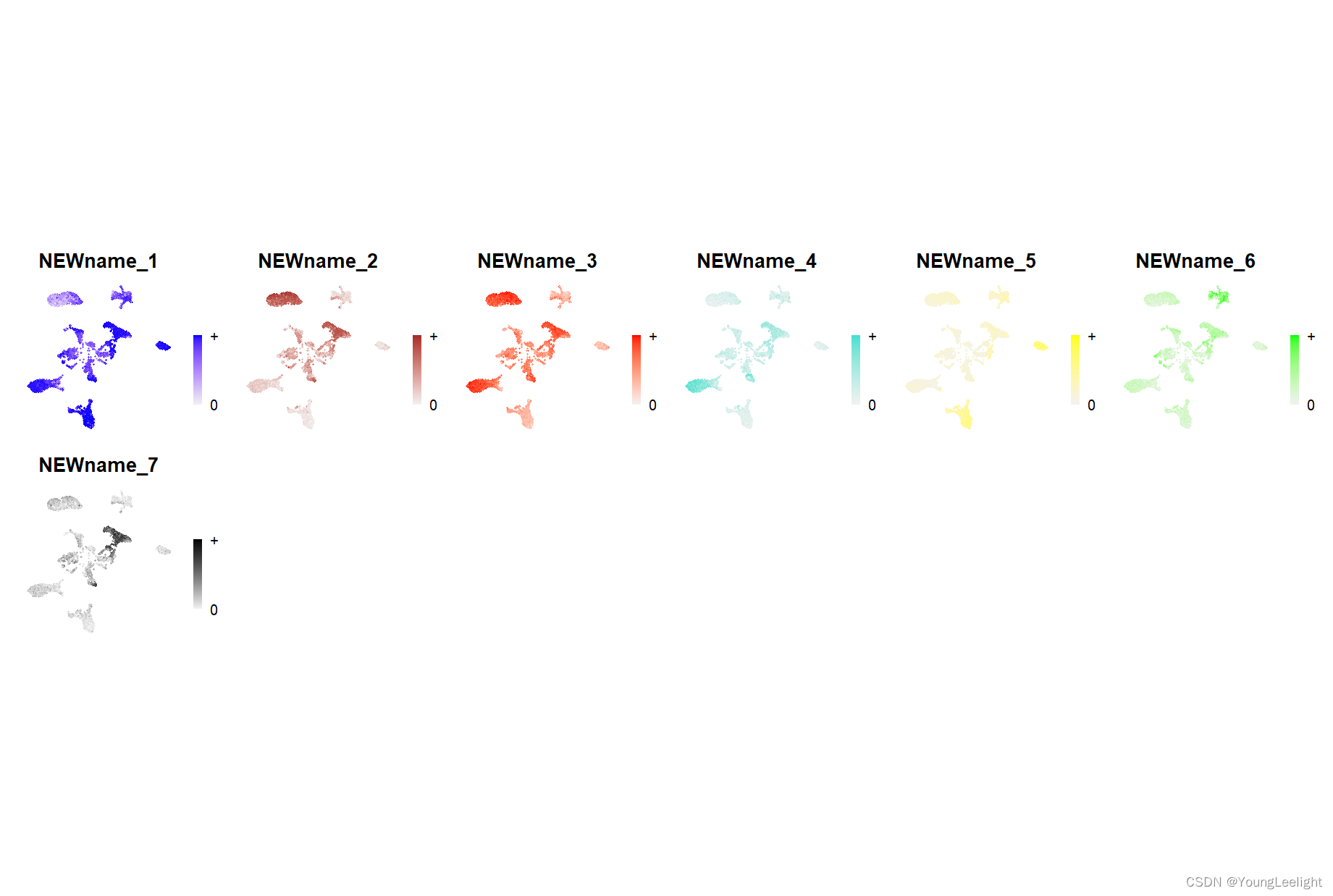
stitch together with patchwork
wrap_plots(plot_list, ncol=6)
4.71 模块间相关性
# plot module correlagram
ModuleCorrelogram(seurat_obj,
exclude_grey = TRUE, # 默认删除灰色模块
features = "hMEs" # What to plot? Can select hMEs, MEs, scores, or average
)
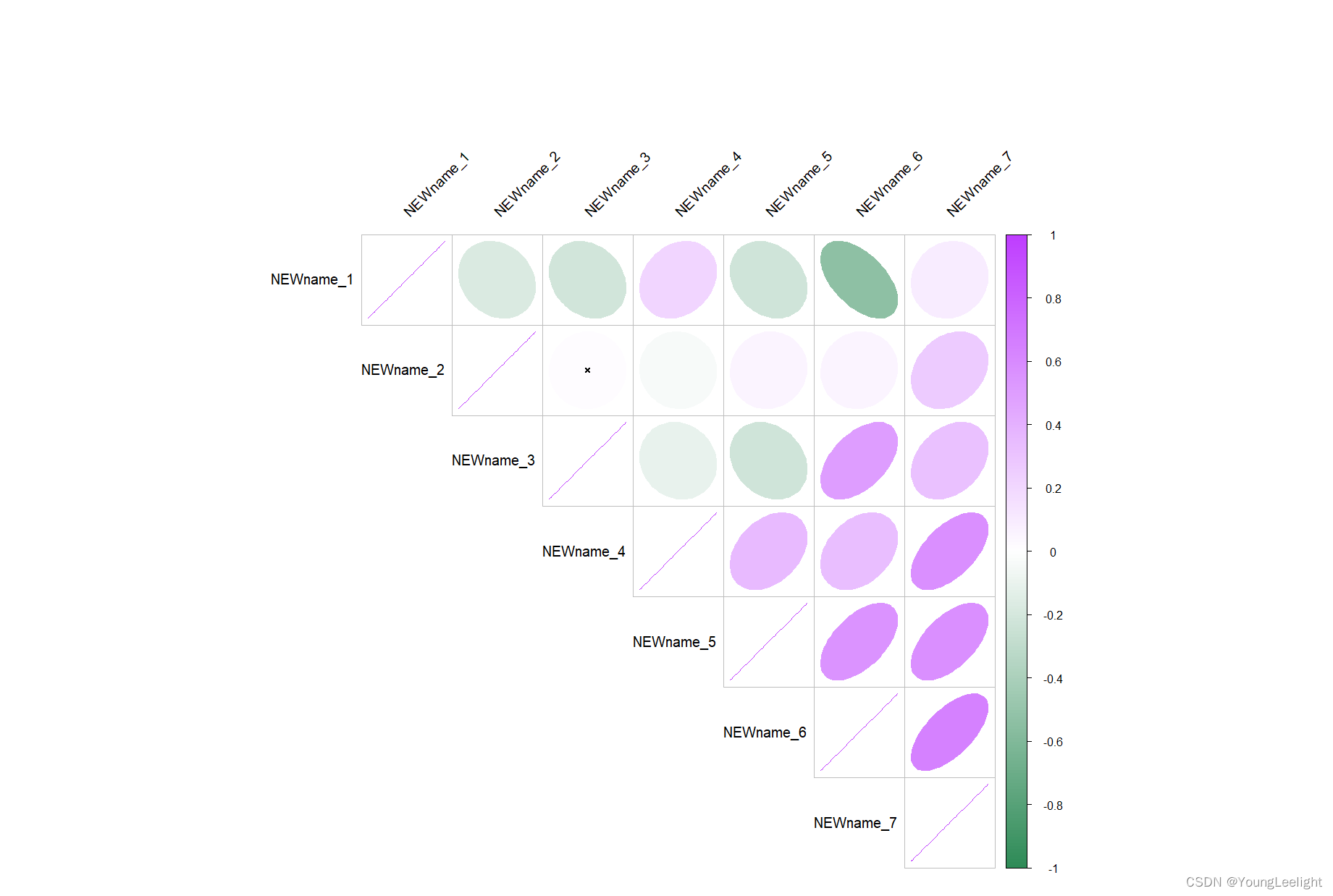
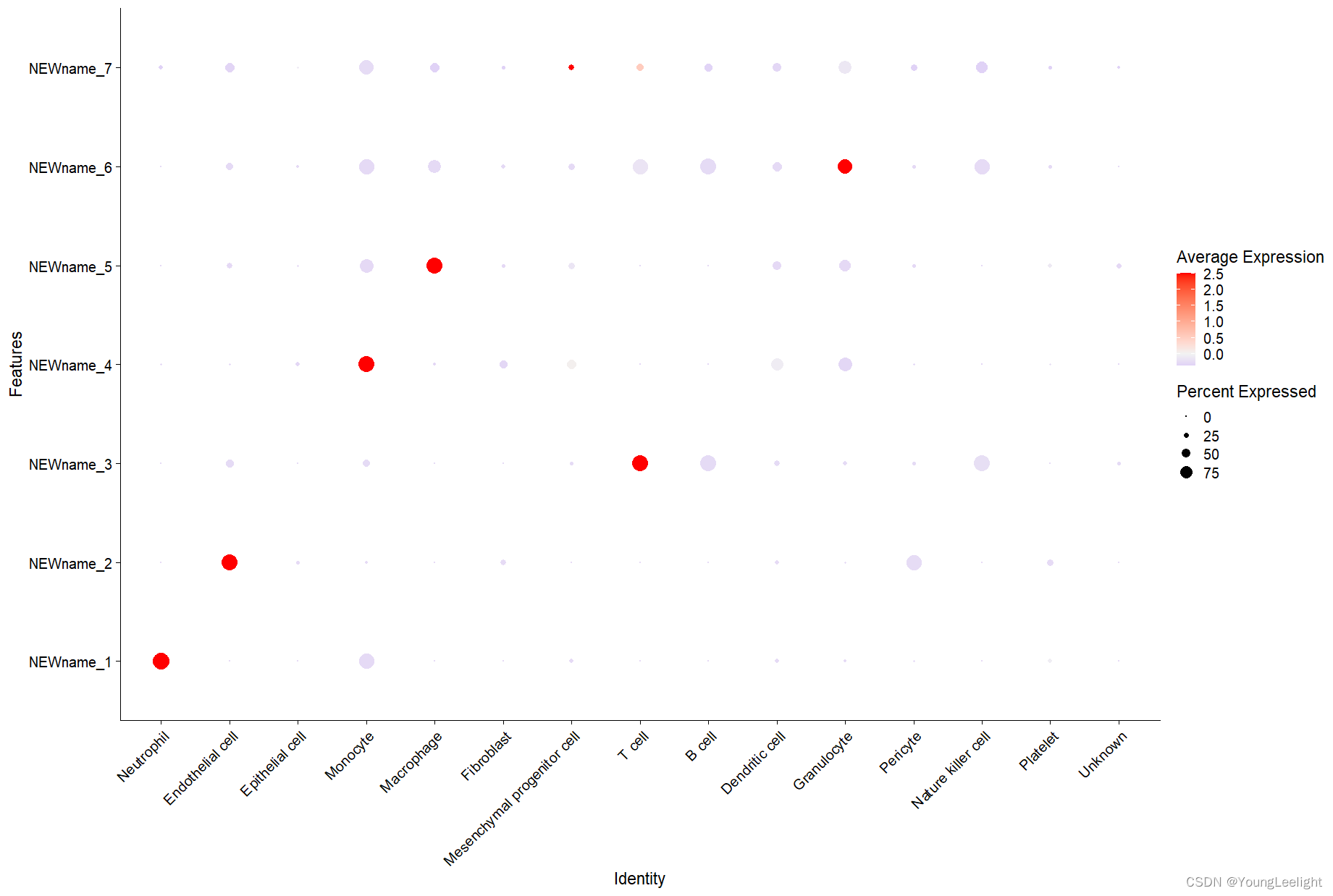
气泡图
# get hMEs from seurat object
MEs <- GetMEs(seurat_obj, harmonized=TRUE)
mods <- colnames(MEs); mods <- mods[mods != 'grey']
# add hMEs to Seurat meta-data:
seurat_obj@meta.data <- cbind(seurat_obj@meta.data, MEs)
# plot with Seurat's DotPlot function
p <- DotPlot(seurat_obj, features=mods, group.by = 'cell_type')
# flip the x/y axes, rotate the axis labels, and change color scheme:
p <- p +
coord_flip() +
RotatedAxis() +
scale_color_gradient2(high='red', mid='grey95', low='blue')
# plot output
p
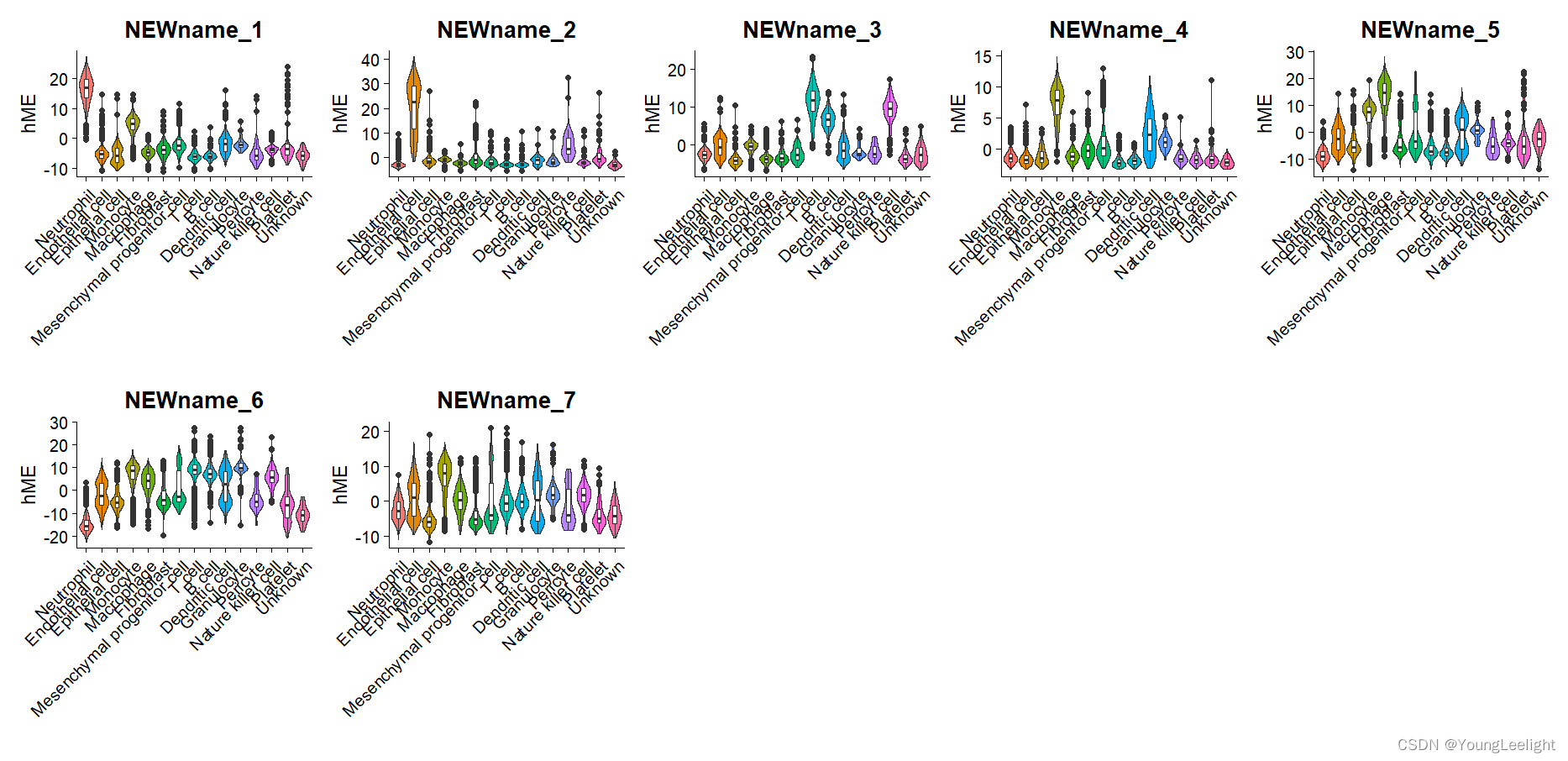
#气泡图
if(T){# Plot INH-M4 hME using Seurat VlnPlot function
p <- VlnPlot(
seurat_obj,
features = 'NEWname_2',
group.by = 'cell_type',
pt.size = 0 # don't show actual data points
)
# add box-and-whisker plots on top:
p <- p + geom_boxplot(width=.25, fill='white')
# change axis labels and remove legend:
p <- p + xlab('') + ylab('hME') + NoLegend()
# plot output
p
}
#批量出图
plot_list <- lapply(mods, function(x) {
print(x)
p <- VlnPlot(
seurat_obj,
features = x,
group.by = 'cell_type',
pt.size = 0 # don't show actual data points
)
# add box-and-whisker plots on top:
p <- p + geom_boxplot(width=.25, fill='white')
# change axis labels and remove legend:
p <- p + xlab('') + ylab('hME') + NoLegend()
p
})
wrap_plots(plot_list, ncol = 5)
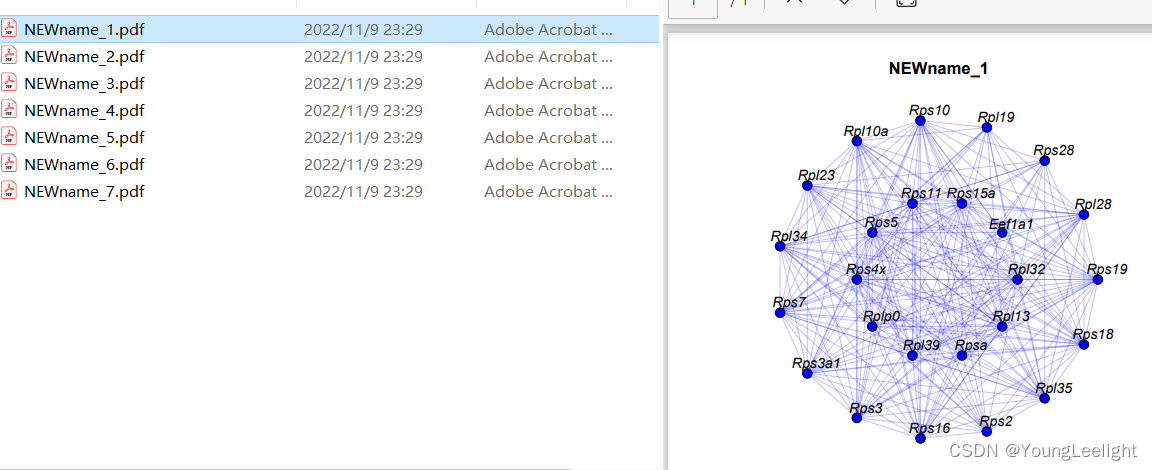
网络可视化
单个模块网络图
library(igraph)
# Visualizes the top hub genes for selected modules as a circular network plot
ModuleNetworkPlot(
seurat_obj,
mods = "all", # all modules are plotted.
outdir = "ModuleNetworks", # The directory where the plots will be stored.
plot_size = c(6, 6),
label_center = FALSE,
edge.alpha = 0.25,
vertex.label.cex = 1, # 基因标签的字体大小
vertex.size = 6 # 节点的大小
)

组合网络图
hubgene network
HubGeneNetworkPlot(
seurat_obj,
mods = "all", # all modules are plotted.
n_hubs = 3,
n_other=6,
edge_prop = 0.75,
)
g <- HubGeneNetworkPlot(seurat_obj, return_graph=TRUE)
#umap网络图
这里使用另一种方法umap来可视化共表达网络中的所有基因,主要在topological overlap matrix (TOM)矩阵计算出umap坐标
seurat_obj <- RunModuleUMAP(
seurat_obj,
n_hubs = 10, # number of hub genes to include for the UMAP embedding
n_neighbors=15, # neighbors parameter for UMAP
min_dist=0.1 # min distance between points in UMAP space
)
# get the hub gene UMAP table from the seurat object
umap_df <- GetModuleUMAP(seurat_obj)
# plot with ggplot
ggplot(umap_df, aes(x=UMAP1, y=UMAP2)) +
geom_point(
color=umap_df$color, # color each point by WGCNA module
size=umap_df$kME*2 # size of each point based on intramodular connectivity
) +
umap_theme()
ModuleUMAPPlot(
seurat_obj,
edge.alpha=0.25,
sample_edges=TRUE,
edge_prop=0.1, # proportion of edges to sample (20% here)
label_hubs=2 ,# how many hub genes to plot per module?
keep_grey_edges=FALSE
)
dev.off()
if(F){
https://github.com/smorabit/hdWGCNA
https://smorabit.github.io/hdWGCNA/articles/basic_tutorial.html
getwd()
file="G:/silicosis/hdgcna"
dir.create(file)
setwd(file)
getwd()
#wget https://swaruplab.bio.uci.edu/public_data/Zhou_2020.rds
#r下载网上的内容
download.file("https://swaruplab.bio.uci.edu/public_data/Zhou_2020.rds","Zhou_2020.rds")
list.files()
1.# single-cell analysis package
library(Seurat)
# plotting and data science packages
library(tidyverse)
library(cowplot)
library(patchwork)
# co-expression network analysis packages:
library(WGCNA)
#devtools::install_github('smorabit/hdWGCNA', ref='dev')
library(hdWGCNA)
# using the cowplot theme for ggplot
theme_set(theme_cowplot())
# set random seed for reproducibility
set.seed(12345)
2# load the Zhou et al snRNA-seq dataset
seurat_obj <- readRDS('Zhou_2020.rds')
#check
p <- DimPlot(seurat_obj, group.by='cell_type', label=TRUE) +
umap_theme() + ggtitle('Zhou et al Control Cortex') + NoLegend()
p
3#Set up Seurat object for WGCNA
length(VariableFeatures(seurat_obj))
names(seurat_obj)
#In this example, we will select genes that are expressed in at least 5% of cells in this dataset, and we will name our hdWGCNA experiment “tutorial”.
seurat_obj <- SetupForWGCNA(
seurat_obj,
gene_select = "fraction", # the gene selection approach
fraction = 0.05, # fraction of cells that a gene needs to be expressed in order to be included
wgcna_name = "tutorial" # the name of the hdWGCNA experiment
)
Assays(seurat_obj)
names(seurat_obj)
seurat_obj@assays
4#metacell
## construct metacells in each group
seurat_obj <- MetacellsByGroups(
seurat_obj = seurat_obj,
group.by = c("cell_type", "Sample"), # specify the columns in seurat_obj@meta.data to group by
k = 25, # nearest-neighbors parameter
max_shared = 10, # maximum number of shared cells between two metacells
ident.group = 'cell_type', # set the Idents of the metacell seurat object
reduction = "harmony"#指定降维方法 不然会报错
)
# normalize metacell expression matrix:
seurat_obj <- NormalizeMetacells(seurat_obj)
rm(ls())
gc()
}




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











