






遇事不决可问春风,春风不语即随本心,可我若是本心坚定,又怎会遇事不决,春风也有春风愁,不牢春风为我忧。
main
from torch.utils.data import DataLoader
import torch
import torch.nn.functional as F
from torch.optim import Adam
from scGNN import GENELink
from torch.optim.lr_scheduler import StepLR
import scipy.sparse as sp
from utils import scRNADataset, load_data, adj2saprse_tensor, Evaluation, Network_Statistic
import pandas as pd
from torch.utils.tensorboard import SummaryWriter
from PytorchTools import EarlyStopping
import numpy as np
import random
import glob
import os
import time
import argparse
parser = argparse.ArgumentParser()#创建一个新的参数解析器对象,用于编写用户友好的命令行接口
parser.add_argument('--lr', type=float, default=3e-3, help='Initial learning rate.')
#为解析器添加命令行参数,每个add_argumet调用定义了一个命令行参数
#lr指定学习率,默认值为3e-3
parser.add_argument('--epochs', type=int, default= 20, help='Number of epoch.')
#设置训练的轮数,默认值为20
parser.add_argument('--num_head', type=list, default=[3,3], help='Number of head attentions.')
#指定多头注意力机制中的头数,默认值为列表[3,3],其中第一个3表示第一阶段的注意力层中包含3个注意力头,
# 第二个3,表示第二阶段的注意力层中包含的3个注意力,第一个阶段可能使用3个头来处理输入序列,而第二个# 阶段可能再次使用另外3个头来进一步处理前一阶段的输出。
parser.add_argument('--alpha', type=float, default=0.2, help='Alpha for the leaky_relu.')
# 指定leak_relu激活函数的负斜率,默认值为0.2,这里详细看附录一
parser.add_argument('--hidden_dim', type=int, default=[128,64,32], help='The dimension of hidden layer')
# 指定隐藏层的维度,详细请看附录二
parser.add_argument('--output_dim', type=int, default=16, help='The dimension of latent layer')
# 指定潜在层的维度,默认值为16,详细看附录3
parser.add_argument('--batch_size', type=int, default=256, help='The size of each batch')
# 用于指定每个批次的大小,默认值为256
parser.add_argument('--loop', type=bool, default=False, help='whether to add self-loop in adjacent matrix')
# 布尔值,用于指定是否在邻接矩阵中添加自环,默认值为false,详细看附录4
parser.add_argument('--seed', type=int, default=8, help='Random seed')
# 指定随机种子,详细看附录5
parser.add_argument('--Type',type=str,default='dot', help='score metric')
#Type:指定评分指标,默认为 'dot'。详细看附录6
parser.add_argument('--flag', type=bool, default=False, help='the identifier whether to conduct causal inference')
# flag:布尔值,标识是否进行因果推断,默认为 False。详细看附录7
parser.add_argument('--reduction',type=str,default='concate', help='how to integrate multihead attention')
# reduction:指定多头注意力机制中如何整合信息,默认为 'concate'(可能是 'concat' 的拼写错误)。详情看附页8
args = parser.parse_args()
# 通过args对象访问这些参数的值,例如args.lr,args.epochs
seed = args.seed
#这行代码从命令行参数中获取一个值,并将其赋值给变量 seed。这个值将用作后续所有随机数生成操作的种
#子。这允许用户在运行脚本时指定一个种子,或者使用默认值(如果用户没有指定)。
random.seed(args.seed)
#这行代码设置了 Python 内置 random 模块的随机种子。这意味着之后使用 random 模块生成的随机数将与种
#子相关联,确保了可重复性。例如,如果你使用 random.randint() 或 random.shuffle() 等函数,设置种
#子后每次运行代码时生成的随机数序列将是相同的。
torch.manual_seed(args.seed)
#这行代码设置了 PyTorch 的随机种子。在 PyTorch 中,很多操作如初始化权重、打乱数据集等都涉及到随机
#数生成。通过设置这个种子,你可以确保每次运行代码时 PyTorch 生成的随机数是相同的。需要注意的是,
#torch.manual_seed() 只影响当前设备(CPU或GPU)。如果你在使用 GPU,可能还需要使用 torch.cuda.manual_seed() 来设置 GPU 的随机种子。
np.random.seed(args.seed)
#这行代码设置了 NumPy 的随机种子。NumPy 也广泛用于数据科学和机器学习中,很多数据处理和生成随机数的#操作都依赖于 NumPy。通过设置这个种子,你可以确保使用 NumPy 生成的随机数(如 np.random.rand() 或 #np.random.normal())在每次运行代码时都是相同的。
#这段Python代码定义了一个函数 embed2file,其目的是将模型学习到的嵌入(embeddings)保存到CSV文件
#中。这些嵌入通常是高维空间中的向量,表示输入数据的某种特征或属性。在这个特定的函数中,tf_embed 和
# tg_embed 分别代表两组不同的嵌入,比如可能表示了转录因子(transcription factors, TFs)和目标基因(target genes)的特征。
def embed2file(tf_embed,tg_embed,gene_file,tf_path,target_path):
#这行代码定义了一个名为 embed2file 的函数,它接受五个参数:
#tf_embed:一个包含转录因子嵌入的张量(Tensor)。
#tg_embed:一个包含目标基因嵌入的张量。
#gene_file:包含基因信息的文件路径,通常是一个CSV文件。
#tf_path:转录因子嵌入保存路径。
#target_path:目标基因嵌入保存路径。
tf_embed = tf_embed.cpu().detach().numpy()
#这行代码将 tf_embed 张量转移到CPU,从计算图中分离(如果它是一个计算图的一部分),并转换为NumPy数
#组。这是必要的步骤,因为NumPy数组是Python中处理数值数据的标准格式,而且CSV文件是用NumPy数组而不是
#张量来保存的。
tg_embed = tg_embed.cpu().detach().numpy()
#类似地,这行代码处理 tg_embed 张量,使其也成为一个NumPy数组。
gene_set = pd.read_csv(gene_file, index_col=0)
#这行代码使用Pandas库读取 gene_file 指定的CSV文件,并将第一列设置为DataFrame的索引。
#这个DataFrame gene_set 包含了基因的信息,其中索引列通常是基因的标识符。
tf_embed = pd.DataFrame(tf_embed,index=gene_set['Gene'].values)
#这行代码创建了一个新的Pandas DataFrame,其数据来自 tf_embed 数组,索引来自 gene_set DataFrame中名为 'Gene' 的列的值。这确保了嵌入向量与其对应的基因标识符匹配。
tg_embed = pd.DataFrame(tg_embed, index=gene_set['Gene'].values)
#这行代码对 tg_embed 数组做同样的处理,创建一个新的DataFrame,其索引也是基于基因标识符。
tf_embed.to_csv(tf_path)
# 这行代码将转录因子的嵌入DataFrame保存到 tf_path 指定的路径。数据将以CSV格式写入文件。
tg_embed.to_csv(target_path)
#这行代码将目标基因的嵌入DataFrame保存到 target_path 指定的路径。同样,数据将以CSV格式写入文件
exp_file = 'F:\\Projects\\GENELink-main\\Dataset\\Benchmark Dataset\\Lofgof Dataset\\mESC\\TFs+500\\BL--ExpressionData.csv'
tf_file = 'F:\\Projects\\GENELink-main\\Dataset\\Benchmark Dataset\\Lofgof Dataset\\mESC\\TFs+500\\TF.csv'
target_file = 'F:\\Projects\\GENELink-main\\Dataset\\Benchmark Dataset\\Lofgof Dataset\\mESC\\TFs+500\\Target.csv'
train_file = 'F:\Projects\GENELink-main\Demo\Train_validation_test\mESC 500\\Train_set.csv'
val_file = 'F:\Projects\GENELink-main\Demo\Train_validation_test\mESC 500\\Validation_set.csv'
tf_embed_path = r'Result/.../Channel1.csv'
target_embed_path = r'Result/.../Channel2.csv'
data_input = pd.read_csv(exp_file,index_col=0)
#data_input = pd.read_csv(exp_file, index_col=0):
#这行代码使用Pandas库从CSV文件中读取数据,并将第一列作为DataFrame的索引。
#exp_file是包含基因表达数据的文件路径。这个步骤通常用于加载原始的单细胞RNA测序数据。
loader = load_data(data_input)
#这行代码创建了一个数据加载器对象loader,它可能是一个自定义的类或函数,
#用于进一步处理和封装基因表达数据。load_data函数可能负责数据的清洗、标准化和其他预处理步骤。
feature = loader.exp_data()
#这行代码调用loader对象的exp_data方法来获取处理后的特征数据。
#这些特征可能包括基因表达水平或其他衍生的统计量,用于后续的模型训练。
tf = pd.read_csv(tf_file,index_col=0)['index'].values.astype(np.int64)
#这行代码加载转录因子(TFs)的数据。它从CSV文件中读取数据,选择index列,并将这些值转换为整数类型(np.int64)。其中index_col=0见详情8
#tf_file是包含转录因子信息的文件路径。这些整数索引可能对应于基因表达矩阵中的行。
target = pd.read_csv(target_file,index_col=0)['index'].values.astype(np.int64)
#类似地,这行代码加载目标基因的数据。它也从CSV文件中读取数据,选择index列,并将这些值转换为整数类型。target_file是包含目标基因信息的文件路径。
feature = torch.from_numpy(feature)
#这行代码将NumPy数组格式的特征数据转换为PyTorch张量。
#这是为了利用PyTorch的计算能力,特别是在GPU上进行高效的张量运算。
tf = torch.from_numpy(tf)
#这行代码将转录因子的索引数组转换为PyTorch张量。
device = torch.device('cuda:0' if torch.cuda.is_available() else 'cpu')
#这行代码设置计算设备。如果系统中有可用的CUDA兼容的GPU,它将使用GPU(cuda:0),
#否则将使用CPU。这是为了确保模型训练和计算可以在最快的可用硬件上执行。
data_feature = feature.to(device)
#这行代码将特征数据移动到指定的计算设备(GPU或CPU)。
tf = tf.to(device)
#这行代码将转录因子的索引数据移动到指定的计算设备。
#总的来说,这段代码负责从文件中读取和预处理数据,将数据转换为适合模型训练的格式,并确保数据被加载到适当的计算设备上
train_data = pd.read_csv(train_file, index_col=0).values
#这行代码使用Pandas库从CSV文件中读取训练数据,并将第一列作为索引。train_file 是训练数据文件的路径。.values 属性将Pandas DataFrame转换为NumPy数组,以便后续处理。
validation_data = pd.read_csv(val_file, index_col=0).values
#类似地,这行代码读取验证数据,并将第一列作为索引。val_file 是验证数据文件的路径。
train_load = scRNADataset(train_data, feature.shape[0], flag=args.flag)
#这行代码创建一个 scRNADataset 数据集对象,它可能是一个自定义类,用于封装单细胞RNA数据集。
#train_data 是训练数据的NumPy数组,feature.shape[0] 指定了特征数量,
#args.flag 是一个命令行参数,用于指示是否进行因果推断。
adj = train_load.Adj_Generate(tf,loop=args.loop)
#这行代码调用 train_load 对象的 Adj_Generate 方法来生成邻接矩阵。
#tf 是转录因子的索引数组,args.loop 是一个命令行参数,用于指示是否在邻接矩阵中添加自环。
adj = adj2saprse_tensor(adj)
#这行代码将邻接矩阵转换为稀疏张量。adj2saprse_tensor 可能是一个自定义函数,用于优化内存使用和计算效率。
train_data = torch.from_numpy(train_data)
#将训练数据的NumPy数组转换为PyTorch张量。
val_data = torch.from_numpy(validation_data)
#将验证数据的NumPy数组转换为PyTorch张量。
#这行代码初始化 GENELink 模型,它是一个用于基因调控网络推断的图神经网络模型。
#模型的参数包括输入维度、隐藏层维度、输出维度、注意力头数、激活函数的负斜率、
#设备(GPU或CPU)、评分指标类型和信息整合方式。这些参数大多数都是从命令行参数中获取的。
model = GENELink(input_dim=feature.size()[1],
hidden1_dim=args.hidden_dim[0],
hidden2_dim=args.hidden_dim[1],
hidden3_dim=args.hidden_dim[2],
output_dim=args.output_dim,
num_head1=args.num_head[0],
num_head2=args.num_head[1],
alpha=args.alpha,
device=device,
type=args.Type,
reduction=args.reduction
)
adj = adj.to(device)
#这行代码将邻接矩阵 adj 移动到指定的计算设备(如GPU)。
#这是必要的步骤,因为模型将在该设备上进行训练,而邻接矩阵是模型训练的一部分。
model = model.to(device)
#这行代码将 GENELink 模型移动到指定的计算设备。
#这样可以确保模型的权重和计算都在同一个设备上进行,从而提高训练效率。
train_data = train_data.to(device)
#这行代码将训练数据移动到指定的计算设备。
validation_data = val_data.to(device)
#这行代码将验证数据移动到指定的计算设备。
optimizer = Adam(model.parameters(), lr=args.lr)
#这行代码创建一个Adam优化器实例,用于模型参数的优化。
#model.parameters() 返回模型中所有需要优化的参数,args.lr 是从命令行参数中获取的学习率。
scheduler = StepLR(optimizer, step_size=1, gamma=0.99)
#这行代码创建一个学习率调度器 StepLR,它属于PyTorch的 torch.optim.lr_scheduler 模块。
#StepLR 调度器会在每 step_size 个epoch后将学习率乘以 gamma。在这个例子中,
#学习率会在每个epoch后乘以0.99,这意味着学习率会以0.99的因子逐渐减小,从而实现学习率衰减。
#
model_path = 'model/...'
if not os.path.exists(model_path):
#这行代码使用 os.path.exists() 函数检查 model_path 指定的路径是否存在。
os.makedirs(model_path)
#如果路径不存在,os.makedirs(model_path) 将创建这个路径及其所有必需的父目录。
#这是一个递归操作,意味着如果路径中的某些中间目录不存在,它们也会被创建。
for epoch in range(args.epochs):
running_loss = 0.0
# 在每个训练周期开始时,初始化 running_loss 变量为 0.0。这个变量通常用于累积计算当前训练周期内的总损失,以便后续计算平均损失或其他相关指标
for train_x, train_y in DataLoader(train_load, batch_size=args.batch_size, shuffle=True):
#使用 DataLoader 从 train_load 数据集中加载训练数据。batch_size 和 shuffle 参数分别控制每个批次的大小和数据是否需要打乱。
model.train()
#model.train() 将模型设置为训练模式。
optimizer.zero_grad()
#optimizer.zero_grad() 清零模型参数的梯度,为新的批次训练准备。
if args.flag:
train_y = train_y.to(device)
else:
train_y = train_y.to(device).view(-1, 1)
#根据 args.flag 的值,将 train_y 标签数据迁移到设备(GPU或CPU)。
# train_y = train_y.to(device).view(-1, 1)
pred = model(data_feature, adj, train_x)
#计算模型的预测输出
#pred = torch.sigmoid(pred)
if args.flag:
pred = torch.softmax(pred, dim=1)
else:
pred = torch.sigmoid(pred)
#根据 args.flag 的值,对预测输出应用softmax或sigmoid激活函数。
loss_BCE = F.binary_cross_entropy(pred, train_y)
#使用二元交叉熵损失函数 F.binary_cross_entropy 计算损失。
loss_BCE.backward()
optimizer.step()
# #loss_BCE.backward() 和 optimizer.step() 进行反向传播和参数更新。
# 向前传播向后传播详细请看附页9
scheduler.step()
running_loss += loss_BCE.item()
#running_loss += loss_BCE.item() 将当前批次的损失累积到 running_loss。
model.eval()
#model.eval() 将模型设置为评估模式。详情看附页10
score = model(data_feature, adj, validation_data)
#使用模型对验证数据进行预测。
if args.flag:
score = torch.softmax(score, dim=1)
else:
score = torch.sigmoid(score)
#这段代码检查 args.flag 的值,并根据这个值应用 softmax 或 sigmoid 激活函数。
#如果 args.flag 为 True,可能表示这是一个多分类任务,因此使用 softmax;
#如果为 False,则可能是二分类任务,因此使用 sigmoid。
#这样的设计允许同一个模型结构适用于不同的任务类型,只需通过命令行参数即可轻松切换。
# score = torch.sigmoid(score)
AUC, AUPR, AUPR_norm = Evaluation(y_pred=score, y_true=validation_data[:, -1],flag=args.flag)
#调用一个自定义的 Evaluation 函数,该函数计算模型在验证数据集上的性能指标。这个函数接受以下参数:
#y_pred=score:模型对验证数据的预测输出。y_true=validation_data[:, -1]:
#验证数据的真实标签,这里选择的是 validation_data DataFrame的最后一列,通常表示为二进制标签(例如,0或1)。
#flag=args.flag:一个命令行参数,可能用于指示是否进行特定的处理,比如因果推断或不同类型的评估逻辑。
#AUC(Area Under the Curve):曲线下面积,通常指的是接收者操作特征曲线(ROC Curve)下的面积,是一个衡量模型分类性能的指标。
#AUPR(Area Under the Precision-Recall Curve):精确率-召回率曲线下的面积,
#是另一个衡量模型性能的指标,尤其在数据类不平衡时更为有效。
#AUPR_norm:可能是 AUPR 的一个规范化版本,用于在不同的数据集或模型间进行比较
print('Epoch:{}'.format(epoch + 1),
'train loss:{}'.format(running_loss),
'AUC:{:.3F}'.format(AUC),
'AUPR:{:.3F}'.format(AUPR))
#Epoch:(epoch + 1):当前的周期编号(从1开始)。
#train loss:(running_loss):当前周期的总训练损失。
#AUC:(AUC):计算得到的 AUC 值,格式化为三位小数。
#AUPR:(AUPR):计算得到的 AUPR 值,格式化为三位小数。
torch.save(model.state_dict(), model_path +'.../.pkl')
#这行代码使用 PyTorch 的 torch.save() 函数将模型的参数(通常称为状态字典或 state_dict)保存到指定的路径。
#model_path +'.../.pkl' 表示保存模型的文件路径和名称,其中 ... 应该被替换为具体的文件名。
#保存模型的状态字典而不是整个模型对象是一种常见做法,因为它允许更灵活地加载模型参数到不同的模型架构中。
model.load_state_dict(torch.load(model_path + model_path +'.../.pkl'))
#这行代码使用 torch.load() 函数从保存的文件中加载模型的状态字典,并使用 model.load_state_dict() 方法将其加载到模型中。
#这样,模型的参数就被恢复到保存时的状态。注意这里的路径字符串 model_path + model_path +'.../.pkl' 可能存在错误,通常应该是 model_path + '.../.pkl'。
model.eval()
#这行代码将模型设置为评估模式,这是在进行模型推理或评估时的标准步骤。
#在评估模式下,模型不会进行权重更新,并且某些特定层(如 dropout 和 batch normalization)的行为会改变,以确保它们在训练和评估时表现一致。
tf_embed, target_embed = model.get_embedding()
#这行代码调用模型的 get_embedding() 方法来获取转录因子(tf)和目标基因(target)的嵌入表示。
#这些嵌入表示通常是模型学习到的低维特征表示,可以用于后续的分析或比较。
embed2file(tf_embed,target_embed,target_file,tf_embed_path,target_embed_path)
#这行代码调用 embed2file() 函数,将转录因子和目标基因的嵌入表示保存到文件中。
#这个函数可能接受嵌入数据和文件路径作为参数,并将这些数据写入到指定的文件中,通常为 CSV 或其他格式,以便于进一步的分析或可视化。
函数运行
附录
附录1 Leaky ReLU
Leaky ReLU(Leaky Rectified Linear Unit)是一种改进版的ReLU(Rectified Linear Unit)激活函数,旨在解决ReLU在训练过程中可能出现的“死亡ReLU”问题。死亡ReLU指的是当输入为负数时,ReLU函数的输出永久地变为零,导致相应的权重参数不再更新。
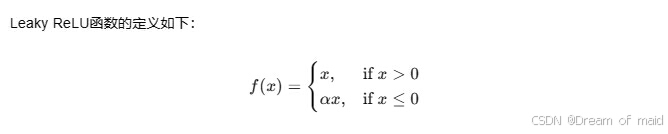
其中,α 是一个很小的正数,通常被称为负斜率(negative slope),它允许负输入有一个非零的梯度。这意味着即使输入是负数,Leaky ReLU也会输出一个非零值,从而允许梯度能够通过,防止神经元死亡。
在你提供的代码中,–alpha 参数就是用来指定Leaky ReLU激活函数的负斜率,默认值为 0.2。这意味着当输入的值小于或等于零时,Leaky ReLU函数将以0.2的斜率输出该值,而不是像标准ReLU那样输出零。
例如,如果输入值为 -2,使用Leaky ReLU激活函数后的输出将是 -0.4(即 -2 * 0.2)。这允许负输入值对模型的训练有所贡献,而不是完全被忽略。
Leaky ReLU相比于ReLU的优点是它可以为负输入值提供非零的梯度,从而使得网络可以学习到更多关于输入数据的信息。然而,Leaky ReLU也有可能导致网络训练不稳定,因为它引入了负输入值的梯度。因此,在使用Leaky ReLU时需要仔细调整负斜率的值,以确保模型的训练效果。
附录2 Hidden_dim
在神经网络中,–hidden_dim 参数用于指定隐藏层的大小或维度。隐藏层是介于输入层和输出层之间的层,它们的目的是在网络中捕捉和学习数据的复杂特征。参数 --hidden_dim 的默认值 [128, 64, 32] 表示网络中包含三层隐藏层,每层的神经元数量分别为 128、64 和 32。
以下是对这个参数的一些详细解释:
第一层隐藏层:这层有 128 个神经元。每个神经元可以看作是一个带有权重和偏置的节点,它接收来自前一层(输入层或前一个隐藏层)的输入,进行加权求和,然后通过激活函数产生输出。
第二层隐藏层:这层有 64 个神经元。它接收来自第一层隐藏层的输出作为输入,并产生自己的输出。
第三层隐藏层:这层有 32 个神经元。它继续接收来自第二层隐藏层的输出,并产生输出,这些输出可能会被传递到下一层,或者作为网络的最终输出。
隐藏层的维度大小对网络的性能有重要影响:
较大的维度:可以增加网络的学习能力,使其能够捕捉更复杂的特征,但也可能导致过拟合和增加计算成本。
较小的维度:可以减少过拟合的风险和计算成本,但可能限制了网络的学习能力。
选择合适的隐藏层维度通常需要基于实验和经验。在实践中,可能需要尝试不同的维度配置,并通过交叉验证等技术来确定最佳的网络结构。
此外,–hidden_dim 参数的值可以根据具体的模型架构和任务需求进行调整。例如,可以使用更少或更多的隐藏层,或者为每个隐藏层选择不同的神经元数量。这种灵活性允许研究人员和工程师根据特定的应用场景定制神经网络的结构。
附录3 output_dim
在机器学习和特别是深度学习中,--output_dim 参数通常用来指定输出层或潜在层(latent layer)的维度。这个参数定义了模型中某个特定层的输出特征的数量。在不同的上下文中,这一层可能有不同的名称,例如潜在层、编码层(在自编码器中)、隐藏层等。对于 --output_dim 参数,默认值 16 表示:
- 维度数量:该层的输出将是一个具有16个元素的向量。在自编码器或神经网络的编码部分,这个向量可能代表输入数据的压缩表示或潜在特征。
- 特征表示:在自动编码器或生成模型中,
--output_dim定义的维度通常用来捕捉数据的核心特征,这些特征应该能够捕捉到数据的本质属性,并且可以用作重建输入数据或生成新数据的基础。 - 降维:在一些应用中,如主成分分析(PCA)或t-SNE,降低数据的维度是为了简化模型的复杂性,提高计算效率,或是为了可视化高维数据。
- 信息瓶颈:在某些模型,如变分自编码器(VAE)中,潜在层的低维表示强制模型学习如何用更少的信息来表示输入数据,这有助于模型学习到更抽象和一般化的特征。
在实际使用中,--output_dim的选择取决于多个因素,包括:
- 数据的复杂性:更复杂的数据可能需要更高的维度来捕捉其特征。
- 模型的容量:更大的模型可能支持更高的潜在维度。
- 计算资源:更高的维度意味着更多的参数,这可能导致更高的计算成本。
- 任务需求:不同的任务可能需要不同程度的特征抽象。
最终,--output_dim的最佳值通常需要通过实验来确定,可能涉及多次迭代和验证,以找到最佳的平衡点。
附录4 LOOP
在图神经网络(Graph Neural Networks, GNNs)和其他处理图结构数据的模型中,--loop 参数通常用来决定是否在图的邻接矩阵中添加自环(self-loops)。自环是一种特殊的边,它从一个节点指向它自己。这个参数的布尔值 False 表示默认情况下不添加自环。
以下是对这个参数的详细解释:
- 自环的作用:在图论中,自环可以增加节点的度数(即与节点相连的边的数量),这可能会影响图的属性和图上运行的算法的行为。在神经网络中,自环允许每个节点在聚合邻居节点的信息时,也考虑自己的特征。
- 图卷积操作:在图卷积网络中,节点的特征更新通常依赖于聚合邻居节点的信息。添加自环意味着在聚合过程中,节点也会考虑自己的特征,这可以增强模型对节点自身特征的感知能力。
- 消息传递:在图神经网络的消息传递过程中,自环允许节点在每次迭代中保留和更新自己的信息。这对于捕获节点的局部结构特征是有益的。
- 模型复杂度:添加自环可能会增加模型的复杂度,因为它引入了额外的信息处理路径。然而,这也提供了额外的能力来捕获和利用节点的自我信息。
- 默认值:
--loop参数的默认值为False,意味着在没有明确指定的情况下,模型不会在邻接矩阵中添加自环。如果你认为自环对于你的任务是有益的,可以将其设置为True。
在实际应用中,是否添加自环取决于具体的任务和图的结构。例如,在社交网络分析中,自环可能表示用户对自己的认知;在生物信息学中,自环可能表示分子自我催化的反应。因此,是否使用自环需要根据具体应用场景和图的特性来决定。
附录5 随机种子
在机器学习和深度学习中,--seed 参数用于指定随机种子。随机种子是一个整数,用于初始化随机数生成器,以确保实验的可重复性和结果的一致性。默认值为 8。
随机种子的作用
- 可重复性:
- 在机器学习模型的训练过程中,许多操作(如权重初始化、数据划分、随机抽样等)都依赖于随机数生成器。使用相同的随机种子可以确保每次运行时生成相同的随机数序列,从而使实验结果可重复。
- 调试和验证:
- 在调试模型时,使用固定的随机种子可以帮助开发者验证模型的行为是否一致,便于定位问题。
- 比较实验:
- 在进行不同模型或超参数的比较时,使用相同的随机种子可以确保所有实验在相同的条件下进行,从而使比较结果更具公正性。
- 避免随机性影响:
- 随机性可能会导致模型性能的波动。通过固定随机种子,可以减少这种波动,使得实验结果更稳定。
如何使用随机种子
在代码中,通常会在初始化随机数生成器时设置随机种子。以下是一些常见的库和如何设置随机种子的示例:
Python 内置的随机库
import random
random.seed(8) # 设置随机种子
NumPy
import numpy as np
np.random.seed(8) # 设置 NumPy 随机种子
TensorFlow
import tensorflow as tf
tf.random.set_seed(8) # 设置 TensorFlow 随机种子
PyTorch
import torch
torch.manual_seed(8) # 设置 PyTorch 随机种子
总结
--seed 参数的设置对于确保实验的可重复性和结果的一致性非常重要。通过在代码中使用固定的随机种子,开发者可以更好地控制实验过程,确保在不同运行之间的结果可比性。
附录6 Type 评定指标或相似度量方法
在机器学习或特定的图神经网络(GNN)模型中,--Type 参数通常用于指定评分指标或相似性度量方法。这个参数决定了模型在预测或评估时使用的打分机制。在你提供的代码中,默认值为 'dot',表示使用的是点积(Dot Product)作为评分指标。
以下是对 --Type 参数的一些详细解释:
- 点积(Dot Product):
- 点积是一种常用的相似性度量方法,特别是在推荐系统和图匹配任务中。两个向量的点积可以衡量它们之间的相似性。如果两个向量的点积较大,表示它们在方向上较为一致,因此相似度较高。
- 其他可能的评分指标:
- 除了点积外,还有其他几种常见的评分指标,例如:
- 余弦相似度(Cosine Similarity):衡量两个向量在方向上的相似性,而不考虑它们的幅度。
- L1/L2 距离:衡量两个向量之间的差异,常用于度量相似性。
- 欧氏距离(Euclidean Distance):衡量两个点在欧几里得空间中的距离。
- 曼哈顿距离(Manhattan Distance):衡量两个点在城市街区中的直线距离。
- 除了点积外,还有其他几种常见的评分指标,例如:
- 自定义评分指标:
- 在某些情况下,你可能需要根据特定任务的需求自定义评分指标。这可能涉及更复杂的数学运算或机器学习模型。
- 参数的应用:
- 在模型训练和预测过程中,
--Type参数的值将决定使用哪种评分函数来评估模型的输出。例如,在图匹配任务中,不同的评分指标可能会对匹配结果产生显著影响。
- 在模型训练和预测过程中,
- 代码实现:
- 在实际代码中,
--Type参数的值将用于选择相应的评分函数。例如,如果使用点积作为评分指标,你可能需要实现或调用一个计算点积的函数。
示例代码片段,展示如何根据--Type参数选择不同的评分函数:
- 在实际代码中,
def dot_product(v1, v2):#点积
return np.sum(v1 * v2)
def cosine_similarity(v1, v2):#余弦相似度
return np.dot(v1, v2) / (np.linalg.norm(v1) * np.linalg.norm(v2))
def evaluate_scores(vectors, type='dot'):# 根据不同的type来选择是点积还是余弦相似度
if type == 'dot':
return dot_product(vectors[0], vectors[1])
elif type == 'cosine':
return cosine_similarity(vectors[0], vectors[1])
else:
raise ValueError("Unsupported scoring type")
# 示例使用
vector1 = np.array([1, 2, 3])
vector2 = np.array([4, 5, 6])
score = evaluate_scores((vector1, vector2), type='dot')
print("Score:", score)
在这个示例中,evaluate_scores 函数根据 --Type 参数的值选择使用点积还是余弦相似度作为评分指标。你可以根据需要扩展这个函数,以支持更多的评分方法。
附录7 flag因果推断
在机器学习模型中,--flag 参数作为一个布尔值,通常用来指示是否启用某个特定的功能或模式。在你提供的代码中,--flag 被用来决定是否进行因果推断,其默认值为 False。
因果推断(Causal Inference)
因果推断是统计学和机器学习中的一个重要领域,它关注的是确定变量之间的因果关系,而不仅仅是相关性。在许多应用中,仅仅了解变量之间的关联是不够的,更重要的是理解一个变量如何影响另一个变量。
使用场景
-
科学研究:在医学、社会科学、经济学等领域,研究者需要确定某些干预措施(如药物、政策变化)是否真正导致了结果的变化。
-
推荐系统:在推荐系统中,因果推断可以帮助我们理解用户行为的变化是否由特定的推荐策略引起。
-
在线广告:在广告效果评估中,因果推断可以用来确定广告曝光是否真正导致了用户的购买行为。
实现方法
在机器学习模型中,进行因果推断可能涉及以下几种方法:
-
随机对照试验(RCT):通过随机分配实验组和对照组来评估干预的效果。
-
观察性研究:使用统计方法来控制混杂变量,从而估计干预的因果效应。
-
工具变量(Instrumental Variables):利用与处理变量相关但与结果变量不直接相关的外部变量来估计因果效应。
-
因果图模型:使用图模型来表示变量之间的因果关系,并使用算法来推断这些关系。
-
机器学习方法:如使用深度学习模型来学习潜在的因果关系。
代码实现
在代码中,--flag 参数可以用来控制是否启用因果推断模式。例如:
import argparse
parser = argparse.ArgumentParser()
parser.add_argument('--flag', type=bool, default=False, help='the identifier whether to conduct causal inference')
args = parser.parse_args()
if args.flag:
# 启用因果推断模式
print("Causal inference mode is enabled.")
# 这里可以添加进行因果推断的代码逻辑
else:
# 禁用因果推断模式
print("Causal inference mode is disabled.")
# 这里可以添加不进行因果推断的代码逻辑
在这个示例中,如果用户在命令行中设置了 --flag 参数(例如,通过运行 python script.py --flag),则模型将启用因果推断模式。否则,将输出提示信息并禁用该模式。
总结
--flag 参数提供了一个灵活的方式来控制模型是否进行因果推断,使得同一个模型可以根据不同的需求进行调整。在实际应用中,根据具体的业务场景和研究目标来决定是否需要进行因果推断。
附页8 多头注意力机制中如何整合信息
在多头注意力机制中,reduction 参数指定了如何整合来自不同头(heads)的信息。默认情况下,这个参数的值通常是 'concat',表示将所有头的输出向量拼接(concatenated)在一起,然后通过一个线性层进行变换,以产生最终的输出。这种方法允许模型在不同的表示子空间中捕捉信息,并将这些信息合并以产生一个综合的表示。
在某些实现中,可能会看到 'reduction' 参数的值被错误地写成了 'concate',这可能是一个拼写错误。正确的形式应该是 'concat'。
多头注意力机制的整合过程通常包括以下步骤:
- 多头计算:对于每个头,计算其对应的注意力分数和加权的值向量。
- 拼接:将所有头的输出向量沿着特征维度拼接在一起。如果每个头的输出维度是
d_v,并且有h个头,那么拼接后的维度将是h * d_v。 - 线性变换:通过一个线性层(通常是全连接层)对拼接后的向量进行变换,以产生最终的输出。这个线性层的权重矩阵通常表示为
W^O,它的维度是h * d_v到d_model,其中d_model是模型的维度。
这种方法的关键在于,它允许模型在不同的头中学习到不同的特征表示,然后将这些表示合并,以增强模型对输入数据的理解。这种机制在 Transformer 架构中尤其重要,因为它使得模型能够捕捉到序列数据中的复杂关系和模式。
在实际应用中,'reduction'参数的设置可能会影响模型的性能和能力。例如,拼接所有头的输出可以增加模型的表达能力,但也可能导致维度过高,从而增加计算负担。因此,根据具体的任务和数据集,可能需要调整这个参数的设置。
附页9 前向传播和反向传播
前向传播是神经网络在进行预测或推断时的数据流向。在这个过程中,输入数据被送入网络,逐层经过加权和激活函数处理,最终在输出层产生预测结果。这个过程可以总结为以下几个步骤:
输入层:输入数据进入网络。
隐藏层:每一层的神经元接收输入,进行加权求和,然后通过激活函数处理,产生输出。
输出层:最后一层神经元的输出作为模型的预测结果。
反向传播(Backpropagation)
反向传播是一种在训练神经网络时使用的算法,用于根据网络的输出误差来计算网络权重的梯度。这个过程是深度学习中的核心,因为它允许网络通过调整权重来最小化损失函数。反向传播包括以下几个步骤:
损失函数计算:首先,根据网络的预测输出和真实标签计算损失函数(如均方误差或交叉熵损失)。
梯度计算:然后,从输出层开始,逆向通过网络的每一层,计算损失函数关于每个权重的梯度。
权重更新:使用这些梯度和梯度下降或其他优化算法来更新网络的权重。
前向传播是数据通过网络的过程,用于生成预测。
反向传播是误差通过网络的过程,用于计算权重的梯度,并更新权重以减少预测误差。
这两个过程在每次训练迭代中交替进行:前向传播生成预测,反向传播根据预测误差调整权重。通过多次迭代这个过程,网络逐渐学习到如何减少其在训练数据上的预测误差。
附页10 训练模式与评估模式
在深度学习中,将模型设置为训练模式(training mode)和评估模式(evaluation mode)是非常重要的,因为这两种模式会影响模型的行为,尤其是在使用某些特定层(如 dropout 和 batch normalization)时。以下是这两种模式的主要区别:
1. 训练模式(Training Mode)
-
激活 Dropout:在训练模式下,Dropout 层会随机“丢弃”一部分神经元,以防止过拟合。这意味着在每次前向传播时,模型的结构会有所不同,从而增强模型的泛化能力。
-
Batch Normalization:在训练模式下,Batch Normalization 层会使用当前批次的均值和方差来标准化数据,并更新运行均值和方差的移动平均值。
-
权重更新:在训练模式下,模型的权重会根据计算出的梯度进行更新。
-
使用
model.train():在 PyTorch 中,使用model.train()将模型设置为训练模式。
2. 评估模式(Evaluation Mode)
-
禁用 Dropout:在评估模式下,Dropout 层会被禁用,所有神经元都会参与计算。这确保了模型在评估时使用全部的特征。
-
使用固定的均值和方差:在评估模式下,Batch Normalization 层会使用在训练过程中计算出的运行均值和方差,而不是当前批次的均值和方差。这确保了评估的一致性。
-
不更新权重:在评估模式下,模型的权重不会更新,通常只进行前向传播以计算输出。
-
使用
model.eval():在 PyTorch 中,使用model.eval()将模型设置为评估模式。
总结
- 训练模式:用于训练模型,启用 Dropout 和 Batch Normalization 的训练行为,允许权重更新。
- 评估模式:用于验证或测试模型,禁用 Dropout 和 Batch Normalization 的训练行为,确保使用固定的统计量进行评估,不更新权重。
在实际应用中,通常在训练过程中交替使用这两种模式:在每个训练周期中使用训练模式进行训练,而在验证或测试阶段使用评估模式来评估模型的性能。这样可以确保模型在实际应用中的表现是可靠的。
utils
import pandas as pd
#Pandas 是一个强大的数据分析和操作库,用于处理结构化数据。pd 是 Pandas 库的常用别名。
import torch
#PyTorch 是一个开源的机器学习库,广泛用于深度学习研究和应用。它提供了强大的GPU加速的张量计算能力。
from torch.utils.data import Dataset
#PyTorch 的 Dataset 类用于定义数据集的抽象类,它规定了如何访问数据集中的数据和标签。
import random as rd
#Python 的 random 模块提供了生成随机数的功能。rd 是 random 模块的别名。
from sklearn.preprocessing import StandardScaler
#Scikit-learn 的 StandardScaler 类用于标准化数据,即从数据中减去平均值并除以标准差,使数据具有零均值和单位方差。
import scipy.sparse as sp
#SciPy 的 scipy.sparse 模块提供了稀疏矩阵的数据结构及其运算,用于高效存储和处理大规模稀疏数据。
import numpy as np
#NumPy 是 Python 中用于科学计算的基础库,提供了多维数组对象和相应的操作。
from sklearn.metrics import roc_auc_score,average_precision_score
#Scikit-learn 的 roc_auc_score 和 average_precision_score 函数用于评估分类模型的性能,
#分别计算接收者操作特征曲线(ROC AUC)和平均精度(AUPR)。
import torch.nn as nn
#PyTorch 的 torch.nn 模块包含了构建神经网络所需的类和函数,如层、激活函数和损失函数。nn 是 torch.nn 模块的常用别名。
class scRNADataset(Dataset):
def __init__(self,train_set,num_gene,flag=False):
super(scRNADataset, self).__init__()
self.train_set = train_set
self.num_gene = num_gene
self.flag = flag
#class scRNADataset(Dataset):定义了一个名为 scRNADataset 的新类,
#它继承自 PyTorch 的 Dataset 类。这使得 scRNADataset 可以被用作 PyTorch 数据加载器(DataLoader)的数据源。
#def __init__(self, train_set, num_gene, flag=False):这是类的初始化方法,它接受三个参数:
#train_set:训练数据集,可能是一个包含基因表达数据的 NumPy 数组或 Pandas DataFrame。
#num_gene:基因的数量,用于后续可能的数据操作或索引。
#flag:一个布尔参数,默认为 False,可能用于控制数据的某些特定处理逻辑。
#super(scRNADataset, self).__init__():这行代码调用父类(Dataset 类)的初始化方法,
#确保 scRNADataset 类正确地继承了 PyTorch Dataset 类的所有属性和方法。
#self.train_set = train_set:将传入的 train_set 参数存储为类的实例变量,以便在类的其他方法中使用。
#self.num_gene = num_gene:将传入的 num_gene 参数存储为类的实例变量。
#最后一个同理
def __getitem__(self, idx):
#这段代码定义了一个 Python 类的 __getitem__ 方法,这个方法通常用在 PyTorch 的 Dataset 类中,
#以支持索引访问数据集中的样本。这个方法允许数据集对象像列表一样通过索引来获取数据和标签。以下是对代码中每个部分的详细解释:
train_data = self.train_set[:,:2]
#这行代码从 self.train_set 中提取所有样本的前两列数据,并将它们存储在 train_data 变量中。这通常表示特征数据。
train_label = self.train_set[:,-1]
#这行代码从 self.train_set 中提取所有样本的最后一列数据,并将它们存储在 train_label 变量中。这通常表示标签数据。
if self.flag:
#这个条件判断用于根据 self.flag 的值决定是否执行特定的数据转换逻辑。
train_len = len(train_label)
#如果 self.flag 为 True,则计算标签的数量并存储在 train_len 变量中。
train_tan = np.zeros([train_len,2])
#创建一个形状为 [train_len, 2] 的零矩阵,并将其存储在 train_tan 变量中。
#这个矩阵将用于存储转换后的标签数据。
train_tan[:,0] = 1 - train_label
#将 train_label 中的每个值从 1 减去并存储在 train_tan 的第一列中。
#这通常用于二分类问题中,将标签转换为一个二维表示,其中一个维度表示负类,另一个维度表示正类。
train_tan[:,1] = train_label
#将原始的 train_label 值存储在 train_tan 的第二列中。
train_label = train_tan
#将转换后的 train_tan 矩阵赋值给 train_label 变量,以替换原始的标签数据。
data = train_data[idx].astype(np.int64)
#使用索引 idx 从 train_data 中获取一个样本的特征数据,并将其数据类型转换为 np.int64。
label = train_label[idx].astype(np.float32)
#使用索引 idx 从 train_label 中获取对应的标签数据,并将其数据类型转换为 np.float32。
return data, label
#返回一个包含特征数据和标签数据的元组
#这个方法允许 PyTorch 的 DataLoader 在训练神经网络时按批次获取数据和标签。通过这种方式,可以高效地加载和处理大规模数据集。
def __len__(self):
return len(self.train_set)
def Adj_Generate(self,TF_set,direction=False, loop=False):
#这段代码定义了一个名为 Adj_Generate 的方法,它是一个用于生成邻接矩阵的方法,通常用于表示基因调控网络中的基因之间的关系。
#这个方法是 scRNADataset 类的一部分,它使用 SciPy 的稀疏矩阵表示来高效地存储和处理大型网络数据。
#TF_set:一个包含转录因子的集合。
#direction:一个布尔参数,用于指示是否考虑基因调控的方向性,默认为 False。
#loop:一个布尔参数,用于指示是否在邻接矩阵中添加自环,默认为 False。
adj = sp.dok_matrix((self.num_gene, self.num_gene), dtype=np.float32)
#这行代码创建一个稀疏的字典键值矩阵(DOK matrix),用于存储邻接矩阵。矩阵的大小由 self.num_gene 决定,数据类型为 np.float32。
for pos in self.train_set:
#这行代码遍历 self.train_set 中的每个元素,其中 self.train_set 可能是一个包含训练数据的列表或数组。
tf = pos[0]
#这行代码从当前位置 pos 中提取第一个元素,即转录因子的索引,并将其存储在 tf 变量中。
target = pos[1]
#这行代码从当前位置 pos 中提取第二个元素,即目标基因的索引,并将其存储在 target 变量中。
if direction == False:
#这个条件判断用于决定是否考虑基因调控的方向性。
if pos[-1] == 1:
#这行代码检查 pos 的最后一个元素是否为 1,如果是,则表示存在一个正向的调控关系。
adj[tf, target] = 1.0
#如果 direction 为 False,则在邻接矩阵中设置 tf 到 target 的边的权重为 1.0。
adj[target, tf] = 1.0
#如果 direction 为 False,则在邻接矩阵中设置 target 到 tf 的边的权重为 1.0,表示双向的调控关系。
else:
if pos[-1] == 1:
adj[tf, target] = 1.0
#如果 direction 为 True,则只设置 tf 到 target 的边的权重为 1.0。
if target in TF_set:
adj[target, tf] = 1.0
#如果 target 也在 TF_set 中,则设置 target 到 tf 的边的权重为 1.0,表示反向的调控关系。
if loop:
adj = adj + sp.identity(self.num_gene)
#这行代码在邻接矩阵中添加单位矩阵,以创建自环。
adj = adj.todok()
#这行代码将 DOK 矩阵转换为 DOK 格式,以便于后续的矩阵运算。
return adj
#返回生成的邻接矩阵。
class load_data():
#它用于加载和预处理数据,包括数据标准化和转换数据类型。
def __init__(self, data, normalize=True):
#这是类的初始化方法,它接受两个参数:
#data:要加载和预处理的数据。
#normalize:一个布尔参数,用于指示是否进行数据标准化,默认为 True。
self.data = data
self.normalize = normalize
def data_normalize(self,data):
#数据标准化方法,接受一个参数data
standard = StandardScaler()
#创建一个 StandardScaler 对象,用于标准化数据。
epr = standard.fit_transform(data.T)
#对数据进行标准化处理,计算每个特征的均值和标准差,并转换数据。
return epr.T
#返回标准化后的数据。
def exp_data(self):
#定义了一个名为 exp_data 的方法,用于获取预处理后的数据。
data_feature = self.data.values
#从实例变量 self.data 中获取数据值。
if self.normalize:
#如果 self.normalize 为 True,则进行数据标准化。
data_feature = self.data_normalize(data_feature)
#调用 data_normalize 方法对数据进行标准化。
data_feature = data_feature.astype(np.float32)
#将数据转换为 np.float32 类型,这是许多机器学习模型中常用的数据类型。
return data_feature
#返回预处理后的数据。
def adj2saprse_tensor(adj):
#这段代码定义了一个名为 adj2saprse_tensor 的函数,它将 SciPy 的稀疏矩阵 adj 转换为 PyTorch 的稀疏张量
#这种转换在处理图数据或网络数据时非常有用,因为稀疏张量可以更高效地表示和操作大型稀疏矩阵。详细请看附录11
coo = adj.tocoo()
#这行代码将输入的稀疏矩阵 adj 转换为 COOrdinate 格式(COO),
#这是一种常见的稀疏矩阵格式,其中矩阵的非零元素及其索引被存储在三个数组中:row、col 和 data。
i = torch.LongTensor([coo.row, coo.col])
#这行代码创建一个 PyTorch 张量 i,它包含 COO 格式矩阵的行索引和列索引。这些索引用于在稀疏张量中定位非零元素。
v = torch.from_numpy(coo.data).float()
#这行代码将 COO 格式矩阵的非零元素数据 coo.data 转换为 PyTorch 张量 v,并确保数据类型为浮点数(float)。
adj_sp_tensor = torch.sparse_coo_tensor(i, v, coo.shape)
#这行代码使用行索引和列索引张量 i、值张量 v 以及原始矩阵的形状 coo.shape 创建一个 PyTorch 稀疏张量 adj_sp_tensor。这个稀疏张量以 COO 格式表示,可以用于后续的图操作和神经网络计算。
return adj_sp_tensor
#返回创建的 PyTorch 稀疏张量 adj_sp_tensor。
def Evaluation(y_true, y_pred,flag=False):
#这段代码定义了一个名为 Evaluation 的函数,它用于计算和返回模型预测的性能指标,
#包括接收者操作特征曲线下面积(AUC)、平均精度(AUPR)和规范化的平均精度(AUPR_norm)。这个函数特别适用于评估分类模型的性能。
#y_true:真实标签的张量。y_pred:模型预测的输出张量。flag:一个布尔参数,用于指示预测输出的处理方式,默认为 False
if flag:
# y_p = torch.argmax(y_pred,dim=1)
y_p = y_pred[:,-1]
#y_p = y_pred[:,-1]:从预测输出中选择最后一列,这通常用于二分类问题,其中模型输出的概率分布的最后一列代表正类的概率。
y_p = y_p.cpu().detach().numpy()
#将预测输出从设备(如GPU)转移到CPU,从计算图中分离,然后转换为NumPy数组。
#从计算图中分离(detach)张量的目的是为了停止追踪该张量在计算图中的梯度。这意味着在反向传播过程中,不会计算和更新与这个张量相关的权重。
y_p = y_p.flatten()
#y_p = y_p.flatten():将数组展平,以便于计算。
else:
y_p = y_pred.cpu().detach().numpy()
#将预测输出从设备转移到CPU,从计算图中分离,然后转换为NumPy数组。
y_p = y_p.flatten()
#将数组展平。
y_t = y_true.cpu().numpy().flatten().astype(int)
#将真实标签从设备转移到CPU,转换为NumPy数组,展平,并转换为整数类型。详细看附录12
AUC = roc_auc_score(y_true=y_t, y_score=y_p)
# roc_auc_score 函数计算接收者操作特征曲线下面积(AUC)
AUPR = average_precision_score(y_true=y_t,y_score=y_p)
#使用 average_precision_score 函数计算平均精度(AUPR)
AUPR_norm = AUPR/np.mean(y_t)
#计算规范化的平均精度(AUPR_norm),通过将AUPR除以真实标签的平均值进行规范化。
return AUC, AUPR, AUPR_norm
def normalize(expression):
std = StandardScaler()
epr = std.fit_transform(expression)
return epr
#这段代码定义了一个名为 normalize 的函数,它使用 StandardScaler 从 scikit-learn 库来标准化输入的数据表达式。
#StandardScaler 是一种常用的数据预处理方法,它通过减去平均值并除以标准差来对数据进行标准化,使得数据符合标准正态分布,即具有零均值和单位方差。
def Network_Statistic(data_type,net_scale,net_type):
#data_type:数据类型,例如 'hESC'、'hHEP' 等。net_scale:网络规模,例如 500 或 1000。
#net_type:网络类型,例如 'STRING'、'Non-Specific'、'Specific' 或 'Lofgof'。
if net_type =='STRING':
dic = {'hESC500': 0.024, 'hESC1000': 0.021, 'hHEP500': 0.028, 'hHEP1000': 0.024, 'mDC500': 0.038,
'mDC1000': 0.032, 'mESC500': 0.024, 'mESC1000': 0.021, 'mHSC-E500': 0.029, 'mHSC-E1000': 0.027,
'mHSC-GM500': 0.040, 'mHSC-GM1000': 0.037, 'mHSC-L500': 0.048, 'mHSC-L1000': 0.045}
query = data_type + str(net_scale)
scale = dic[query]
return scale
#这里定义了一个字典 dic,其中包含了不同数据类型和网络规模的统计值。通过将 data_type 和 net_scale 拼接成字符串 query,在字典中查找相应的值并返回
elif net_type == 'Non-Specific':
dic = {'hESC500': 0.016, 'hESC1000': 0.014, 'hHEP500': 0.015, 'hHEP1000': 0.013, 'mDC500': 0.019,
'mDC1000': 0.016, 'mESC500': 0.015, 'mESC1000': 0.013, 'mHSC-E500': 0.022, 'mHSC-E1000': 0.020,
'mHSC-GM500': 0.030, 'mHSC-GM1000': 0.029, 'mHSC-L500': 0.048, 'mHSC-L1000': 0.043}
query = data_type + str(net_scale)
scale = dic[query]
return scale
elif net_type == 'Specific':
dic = {'hESC500': 0.164, 'hESC1000': 0.165,'hHEP500': 0.379, 'hHEP1000': 0.377,'mDC500': 0.085,
'mDC1000': 0.082,'mESC500': 0.345, 'mESC1000': 0.347,'mHSC-E500': 0.578, 'mHSC-E1000': 0.566,
'mHSC-GM500': 0.543, 'mHSC-GM1000': 0.565,'mHSC-L500': 0.525, 'mHSC-L1000': 0.507}
query = data_type + str(net_scale)
scale = dic[query]
return scale
elif net_type == 'Lofgof':
dic = {'mESC500': 0.158, 'mESC1000': 0.154}
query = 'mESC' + str(net_scale)
scale = dic[query]
return scale
else:
raise ValueError
#如果 net_type 的值不在预期的范围内,函数会抛出一个 ValueError 错误。附录13
附录11
Sparse Tensor:
tensor(indices=tensor([[0, 1, 2],
[2, 0, 1]]), values=tensor([4., 5., 6.]), size=(3, 3))
Dense Version:
tensor([[0., 0., 0.],
[0., 0., 5.],
[6., 0., 4.]])
在这个例子中,稀疏张量表示一个 3x3 的矩阵,其中只有三个非零元素:(0, 2) 位置的元素为 4,(1, 0) 位置的元素为 5,(2, 1) 位置的元素为 6。
使用稀疏张量可以显著减少内存使用,特别是在处理大规模稀疏数据时。此外,PyTorch 还提供了其他稀疏张量操作,如稀疏矩阵乘法、索引和切片等,这些操作都是针对稀疏数据优化的。
附录12
-
从设备转移到CPU:
- 在使用GPU进行训练时,数据(包括模型参数和输入输出数据)通常存储在GPU的内存中以提高计算速度。
- 但是,某些操作,如计算性能指标(如准确度、精确度、召回率等),需要在CPU上进行,因为这些操作可能涉及与外部库的交互,而这些库可能不支持GPU操作。
- 使用
.cpu()方法将数据从GPU内存转移到主机内存(即CPU内存)。
-
转换为NumPy数组:
- NumPy是Python中用于科学计算的基础库,提供了强大的多维数组对象和相应的操作。
- 某些评估指标的计算可能需要使用NumPy库,因为它提供了广泛的数学和统计函数。
- 使用
.numpy()方法将PyTorch张量转换为NumPy数组。
-
展平(Flatten):
- 性能评估函数通常需要一维数组作为输入,因为它们按顺序处理每个样本。
- 如果原始标签是多维的(例如,批量数据),需要将它们展平为一维数组以便于处理。
- 使用
.flatten()方法将多维数组转换为一维数组。
-
转换为整数类型:
- 真实标签通常表示类别索引,这些索引应该是整数。
- 在计算某些性能指标时,整数类型的标签是必需的,因为它们用于确定样本是否属于特定的类别。
- 使用
.astype(int)方法将NumPy数组的数据类型转换为整数。
附录13
在这段代码中,不同的网络类型(net_type)对应于不同的操作,主要是因为每种网络类型可能具有不同的统计特性和数据集,因此需要不同的处理逻辑和参数。以下是为什么对不同网络类型进行不同操作的几个原因:
-
数据集差异:
不同的网络类型可能基于不同的数据集构建,这些数据集在大小、密度、节点分布等方面可能有很大差异。例如,STRING网络可能基于蛋白质-蛋白质相互作用数据,而Non-Specific网络可能基于转录组数据。因此,每种网络类型需要根据其数据集的特性进行特定的统计计算。 -
统计特性差异:
每种网络类型可能具有不同的统计特性,如节点的度分布、聚类系数、路径长度等。这些特性可能影响网络分析的结果,因此需要根据网络类型选择适当的统计方法和参数。 -
应用场景差异:
不同的网络类型可能用于不同的应用场景,如疾病相关基因网络、蛋白质相互作用网络、社交网络等。不同场景下,网络的结构和功能可能有很大差异,因此需要针对特定场景进行定制化的分析。 -
分析目的差异:
对于不同的网络类型,分析的目的可能不同。例如,某些网络可能更关注于识别关键节点或模块,而另一些网络可能更关注于预测节点间的相互作用。因此,需要根据分析目的选择合适的网络类型和相应的操作。 -
计算效率和资源限制:
不同网络类型的规模和复杂度可能不同,这可能影响计算效率和资源消耗。例如,大规模网络可能需要更高效的算法和更多的计算资源。因此,针对不同网络类型进行优化,可以提高分析的效率和可扩展性。 -
预定义参数和阈值:
在代码中,每种网络类型可能已经预定义了一系列参数和阈值,这些参数和阈值是根据以往的研究和实验确定的。使用这些预定义的值可以确保分析结果的一致性和可比性。
总之,对不同网络类型进行不同的操作是为了确保分析的准确性、有效性和适用性。通过针对每种网络类型的特点进行定制化的处理,可以更好地揭示网络的结构和功能,从而为后续的生物学或网络科学研究提供有力的支持。
scGNN
import pandas as pd
import torch
#PyTorch是一个开源的机器学习库,广泛用于深度学习研究和应用。它提供了强大的GPU加速的张量计算能力。
import torch.nn as nn
#PyTorch的torch.nn模块包含了构建神经网络所需的类和函数,如层、激活函数和损失函数。nn是torch.nn模块的常用别名。
import torch.nn.functional as F
#PyTorch的torch.nn.functional模块包含了构建神经网络所需的函数形式的接口,
#这些接口提供了一些额外的函数,它们在构建自定义层或损失函数时非常有用。F是torch.nn.functional模块的常用别名。
import torch.optim as optm
#PyTorch的torch.optim模块包含了多种优化算法,用于在训练神经网络时更新模型的权重。optm是torch.optim模块的常用别名。
from torch.nn import CosineSimilarity
#这行代码从torch.nn模块中导入了CosineSimilarity类,这个类用于计算两个张量之间的余弦相似度,这在一些机器学习任务中,如推荐系统或聚类分析中,是一个非常有用的度量。
class GENELink(nn.Module):
#代码定义了一个名为 GENELink 的 PyTorch 模型类,它用于基因调控网络(Gene Regulatory Network, GRN)的链接预测。
#这个模型基于图注意力网络(Graph Attention Network, GAT),能够学习基因的低维表示,并预测潜在的调控关系。
def __init__(self,input_dim,hidden1_dim,hidden2_dim,hidden3_dim,output_dim,num_head1,num_head2,
alpha,device,type,reduction):
super(GENELink, self).__init__()
self.num_head1 = num_head1
self.num_head2 = num_head2
self.device = device
self.alpha = alpha
self.type = type
self.reduction = reduction
# 这些行设置了模型的各种属性,包括注意力头的数量、设备、激活函数的负斜率、模型类型和数据整合方式。
if self.reduction == 'mean':
self.hidden1_dim = hidden1_dim
self.hidden2_dim = hidden2_dim
elif self.reduction == 'concate':
self.hidden1_dim = num_head1*hidden1_dim
self.hidden2_dim = num_head2*hidden2_dim
#根据 reduction 参数的值,计算第一层和第二层隐藏层的维度。如果 reduction 是 'concate',则将每个注意力头的输出维度相加。
self.ConvLayer1 = [AttentionLayer(input_dim,hidden1_dim,alpha) for _ in range(num_head1)]
#这行代码创建了一个包含 num_head1 个 AttentionLayer 实例的列表。
#每个 AttentionLayer 接受输入维度 input_dim,输出维度 hidden1_dim,以及激活函数的负斜率 alpha。
#列表推导式 [AttentionLayer(input_dim, hidden1_dim, alpha) for _ in range(num_head1)] 根据给定的参数生成指定数量的注意力层。
#通过这种方式,模型的 GENELink 类构建了第一层图注意力层,这些层将用于学习输入数据的低维表示。
#每个注意力头可以独立地学习输入数据的不同特征,并将这些特征组合起来,以捕捉更复杂的模式和关系。
for i, attention in enumerate(self.ConvLayer1):
#将注意力层添加到模型
self.add_module('ConvLayer1_AttentionHead{}'.format(i),attention)
#这行代码遍历 self.ConvLayer1 列表中的每个 AttentionLayer 实例,并使用 self.add_module 方法将它们添加到模型中。
#enumerate(self.ConvLayer1) 函数返回每个层的索引 i 和层对象 attention。
#self.add_module 方法将每个层添加到模型的子模块中,其中第一个参数是子模块的名称
#(例如 'ConvLayer1_AttentionHead0'、'ConvLayer1_AttentionHead1' 等),第二个参数是层对象本身。
self.ConvLayer2 = [AttentionLayer(self.hidden1_dim,hidden2_dim,alpha) for _ in range(num_head2)]
#[AttentionLayer(self.hidden1_dim, hidden2_dim, alpha) for _ in range(num_head2)] 根据给定的参数生成指定数量的注意力层。
for i, attention in enumerate(self.ConvLayer2):
self.add_module('ConvLayer2_AttentionHead{}'.format(i),attention)
#这行代码遍历 self.ConvLayer2 列表中的每个 AttentionLayer 实例,并使用 self.add_module 方法将它们添加到模型中。
#enumerate(self.ConvLayer2) 函数返回每个层的索引 i 和层对象 attention。self.add_module 方法将每个层添加到模型的子模块中,其中第一个参数是子模块的名称(例如 'ConvLayer2_AttentionHead0'、'ConvLayer2_AttentionHead1' 等),第二个参数是层对象本身。
#通过这种方式,模型的 GENELink 类构建了第二层图注意力层,这些层将用于进一步处理第一层注意力层的输出,学习更高层次的特征表示。
#每个注意力头可以独立地学习输入数据的不同特征,并将这些特征组合起来,以捕捉更复杂的模式和关系。
#这段代码定义了 GENELink 类中的四组线性层,用于处理转录因子和目标基因的特征
self.tf_linear1 = nn.Linear(hidden2_dim,hidden3_dim)
#self.tf_linear1 是一个线性层,用于将转录因子的特征从 hidden2_dim 维度转换为 hidden3_dim 维度。
self.target_linear1 = nn.Linear(hidden2_dim,hidden3_dim)
#self.target_linear1 是另一个线性层,用于将目标基因的特征从 hidden2_dim 维度转换为 hidden3_dim 维度。
self.tf_linear2 = nn.Linear(hidden3_dim,output_dim)
#self.tf_linear2 是一个线性层,用于将转录因子的特征从 hidden3_dim 维度转换为 output_dim 维度。
self.target_linear2 = nn.Linear(hidden3_dim, output_dim)
#self.target_linear2 是另一个线性层,用于将目标基因的特征从 hidden3_dim 维度转换为 output_dim 维度。
#这些线性层是全连接层,它们在神经网络中用于特征转换和分类。在 GENELink 模型中,
#这些层用于将图注意力层的输出转换为最终的嵌入表示,这些嵌入表示可以用于后续的链接预测任务。
#第一组线性层(tf_linear1 和 target_linear1)通常用于将注意力层的输出转换为中间特征表示,
#而第二组线性层(tf_linear2 和 target_linear2)用于将这些中间特征表示转换为最终的预测输出。
#这种结构允许模型在不同的层次上捕捉和学习数据的特征,从而提高链接预测的准确性。
if self.type == 'MLP':
self.linear = nn.Linear(2*output_dim, 2)
#self.linear 是一个线性层(全连接层),它将两个输入特征(每个特征维度为 output_dim)拼接后的维度(即 2 * output_dim)映射到输出维度为 2 的空间。
#这通常用于二分类任务,其中输出层的两个单元代表两个类别的预测概率。
#nn.Linear 是 PyTorch 中定义线性层的类,其第一个参数是输入特征的维度,第二个参数是输出特征的维度
self.reset_parameters()
#调用 reset_parameters 方法来初始化模型中所有可学习的参数(权重和偏置)。
#这个方法在类定义中应该已经实现,通常用于应用特定的初始化策略,例如 Xavier 初始化或零初始化,以促进模型训练的稳定性和收敛性
#简而言之,这段代码根据模型类型决定是否添加一个额外的线性层,用于处理特定类型的任务(如二分类),并重置模型的所有参数以准备训练。
def reset_parameters(self):
for attention in self.ConvLayer1:
attention.reset_parameters()
#这行代码遍历 self.ConvLayer1 列表中的每个 AttentionLayer 实例,并调用它们的 reset_parameters 方法。这通常用于重置注意力层的权重和偏置。
for attention in self.ConvLayer2:
attention.reset_parameters()
#这行代码遍历 self.ConvLayer2 列表中的每个 AttentionLayer 实例,并调用它们的 reset_parameters 方法。
nn.init.xavier_uniform_(self.tf_linear1.weight,gain=1.414)
#使用 PyTorch 的 nn.init 模块中的 xavier_uniform_ 函数来初始化 self.tf_linear1 线性层的权重。
#xavier_uniform_ 是一种流行的权重初始化方法,它根据前一层的维度来调整权重的方差。gain=1.414 是一个缩放因子,用于控制初始化的方差。
nn.init.xavier_uniform_(self.target_linear1.weight, gain=1.414)
#使用 xavier_uniform_ 方法初始化 self.target_linear1 线性层的权重。
nn.init.xavier_uniform_(self.tf_linear2.weight, gain=1.414)
nn.init.xavier_uniform_(self.target_linear2.weight, gain=1.414)
def encode(self,x,adj):
#代码定义了 GENELink 类中的 encode 方法,该方法用于对输入特征数据 x 和邻接矩阵 adj 进行编码
if self.reduction =='concate':
x = torch.cat([att(x, adj) for att in self.ConvLayer1], dim=1)
#这行代码遍历 self.ConvLayer1 中的所有注意力层,对每个注意力层调用 att(x, adj) 来获取输出。
#然后,使用 torch.cat 将所有输出在维度 1(特征维度)上拼接起来。这意味着每个注意力头的输出将被拼接成一个更长的特征向量。
x = F.elu(x)
#对拼接后的特征向量 x 应用 ELU(Exponential Linear Unit)激活函数。ELU 是一种激活函数,它可以为负输入提供非零的输出,有助于缓解梯度消失问题
elif self.reduction =='mean':
x = torch.mean(torch.stack([att(x, adj) for att in self.ConvLayer1]), dim=0)
#这行代码遍历 self.ConvLayer1 中的所有注意力层,对每个注意力层调用 att(x, adj) 来获取输出
#。然后,使用 torch.stack 将所有输出在维度 0(批次维度)上堆叠起来,接着计算这些输出的均值。这意味着每个注意力头的输出将被平均,以得到一个单一的特征向量。
x = F.elu(x)
else:
raise TypeError
out = torch.mean(torch.stack([att(x, adj) for att in self.ConvLayer2]),dim=0)
#这个列表推导式遍历 self.ConvLayer2 中的所有注意力层对象,并对每个注意力层调用 att(x, adj) 方法。
#这里 x 是输入的特征数据,adj 是邻接矩阵。对于每个注意力层,这将产生一个输出特征矩阵。
#torch.stack 函数将上述列表推导式生成的所有输出特征矩阵沿着新的维度 0(通常是批次维度)堆叠起来。
#这意味着如果 self.ConvLayer2 中有 num_head2 个注意力层,那么将有 num_head2 个输出特征矩阵被堆叠成一个多维张量。
#torch.mean 函数计算堆叠后的张量沿着维度 0 的均值。
#由于 torch.stack 生成了一个形状为 (num_head2, N, hidden2_dim) 的张量(其中 N 是节点的数量,hidden2_dim 是每个注意力头的输出维度),
#计算均值后将得到一个形状为 (N, hidden2_dim) 的张量,其中每个节点的特征是所有注意力头输出的平均值。
return out
#最终,计算得到的均值张量被赋值给变量 out,它代表了第二层注意力层的编码输出,可以用于后续的模型操作,如链接预测或特征表示。
def decode(self,tf_embed,target_embed):
#def decode(self, tf_embed, target_embed): 定义了一个名为 decode 的方法,它接受三个参数:
#self(指向类实例的引用),tf_embed(目标函数的嵌入向量),和 target_embed(目标的嵌入向量)。
if self.type =='dot':
prob = torch.mul(tf_embed, target_embed)
#prob = torch.mul(tf_embed, target_embed) 使用 PyTorch 的 mul 函数计算两个嵌入向量的逐元素乘积。
prob = torch.sum(prob,dim=1).view(-1,1)
#prob = torch.sum(prob, dim=1).view(-1, 1) 计算乘积向量的元素和(沿着维度1,即每个向量的元素和),然后将结果重塑为一个二维张量,其中每行是一个概率值。
return prob
#返回计算出的概率
elif self.type =='cosine':
prob = torch.cosine_similarity(tf_embed,target_embed,dim=1).view(-1,1)
# 使用 PyTorch 的 cosine_similarity 函数计算两个嵌入向量之间的余弦相似度,然后重塑结果为二维张量。
return prob
elif self.type == 'MLP':
h = torch.cat([tf_embed, target_embed],dim=1)
#使用 PyTorch 的 cat 函数将两个嵌入向量沿着维度1(特征维度)连接起来。
prob = self.linear(h)
#将连接后的向量通过一个线性层(可能是类的成员变量)来计算概率。
return prob
else:
raise TypeError(r'{} is not available'.format(self.type))
def forward(self,x,adj,train_sample):
#它是一个深度学习模型中的前向传播函数。这个方法接受三个参数:
#self(指向类实例的引用),x(节点的特征矩阵),adj(邻接矩阵,用于表示图结构中节点之间的关系),以及 train_sample(训练样本索引的矩阵)。
#这个方法的目的是计算模型的预测输出
embed = self.encode(x,adj)
#embed = self.encode(x, adj):调用模型的 encode 方法来编码输入的特征矩阵 x 和邻接矩阵 adj,得到节点的嵌入表示 embed。
tf_embed = self.tf_linear1(embed)
#tf_embed = self.tf_linear1(embed):将编码后的嵌入通过第一个线性变换层 tf_linear1 进行变换。
tf_embed = F.leaky_relu(tf_embed)
#对变换后的嵌入应用 LeakyReLU 激活函数,这是一种改进的ReLU激活函数,允许负值有一个非零的梯度。
tf_embed = F.dropout(tf_embed,p=0.01)
#对激活后的嵌入应用 dropout,这是一种正则化技术,用于防止过拟合。p=0.01 表示随机丢弃1%的节点。
tf_embed = self.tf_linear2(tf_embed)
#将dropout后的嵌入通过第二个线性变换层 tf_linear2 进行变换
tf_embed = F.leaky_relu(tf_embed)
#再次应用 LeakyReLU 激活函数。
target_embed = self.target_linear1(embed)
target_embed = F.leaky_relu(target_embed)
target_embed = F.dropout(target_embed, p=0.01)
target_embed = self.target_linear2(target_embed)
target_embed = F.leaky_relu(target_embed)
self.tf_ouput = tf_embed
self.target_output = target_embed
#将变换后的目标函数嵌入和目标嵌入保存为类的属性,可能用于后续的计算或分析。
train_tf = tf_embed[train_sample[:,0]]
train_target = target_embed[train_sample[:, 1]]
#矩阵中的索引,从目标函数嵌入和目标嵌入中选择对应的训练样本。
pred = self.decode(train_tf, train_target)
#调用 decode 方法,传入选择的训练样本,计算预测输出。
return pred
def get_embedding(self):
return self.tf_ouput, self.target_output
class AttentionLayer(nn.Module):
#这段代码定义了一个名为 AttentionLayer 的类,它是一个神经网络模块,继承自 PyTorch 的 nn.Module。
#这个类实现了一个注意力层,它可以被用来加权输入特征,以便模型可以关注输入序列中更重要的部分
def __init__(self,input_dim,output_dim,alpha=0.2,bias=True):
#self:指向类实例的引用。input_dim:输入特征的维度。output_dim:输出特征的维度。
#alpha:一个超参数,用于控制注意力机制的平滑程度,默认值为0.2。bias:一个布尔值,指示是否在注意力机制中包含偏置项,默认为True。
super(AttentionLayer, self).__init__()
self.input_dim = input_dim
self.output_dim = output_dim
self.alpha = alpha
self.weight = nn.Parameter(torch.FloatTensor(self.input_dim, self.output_dim))
#定义一个可学习的权重矩阵,其维度为 input_dim x output_dim。
self.weight_interact = nn.Parameter(torch.FloatTensor(self.input_dim,self.output_dim))
#定义另一个可学习的权重矩阵,用于计算注意力交互。
self.a = nn.Parameter(torch.zeros(size=(2*self.output_dim,1)))
#定义一个可学习的参数向量 a,其维度为 2*output_dim x 1,初始值为零。
if bias:
self.bias = nn.Parameter(torch.FloatTensor(self.output_dim))
#定义一个可学习的偏置向量。
else:
self.register_parameter('bias', None)
#注册一个名为 'bias' 的参数,但不创建它(即设置为None)。
self.reset_parameters()
#调用一个方法来初始化模块的参数。这个方法没有在这段代码中定义,
#但它应该在类的其他部分定义,用于设置权重和偏置的初始值。
def reset_parameters(self):
#这个方法的目的是初始化模块的权重和偏置参数,通常在模块创建时被调用
nn.init.xavier_uniform_(self.weight.data, gain=1.414)
#使用 Xavier 初始化方法(也称为 Glorot 初始化)来初始化权重矩阵 self.weight。Xavier 初始化是一种流行的权重初始化方法,
#它考虑到了前一层和后一层的维度,以保持激活函数输入的方差在传播过程中的一致性。gain=1.414 是一个缩放因子,
#通常设置为输入和输出维度的乘积的平方根(在这个例子中,由于是均匀分布,所以使用1.414,即根号2)。
nn.init.xavier_uniform_(self.weight_interact.data, gain=1.414)
#:同样使用 Xavier 初始化方法来初始化另一个权重矩阵 self.weight_interact。
if self.bias is not None:
self.bias.data.fill_(0)
#如果存在偏置项,则将其所有元素初始化为0。这是通过 fill_ 方法实现的,它会将张量中的所有元素设置为指定的值。
nn.init.xavier_uniform_(self.a.data, gain=1.414)
#使用 Xavier 初始化方法来初始化参数向量 self.a。
#这个方法确保了在模型训练开始之前,所有的权重和偏置都被赋予了合适的初始值。
#合理的初始化对于深度学习模型的训练非常重要,因为它可以帮助模型更快地收敛,
#并且减少梯度消失或爆炸的风险。在这段代码中,权重被初始化为均匀分布,而偏置则被初始化为0
def _prepare_attentional_mechanism_input(self, x):
#它是 AttentionLayer 类的一个辅助方法,用于准备注意力机制的输入。
#这个方法接受一个参数 self,指向类的实例,以及 x,表示输入数据
Wh1 = torch.matmul(x, self.a[:self.output_dim, :])
#计算输入数据 x 和参数向量 self.a 的前半部分的矩阵乘积。这里 self.a[:self.output_dim, :] 表示选取 self.a 的前 output_dim 行,保持列不变。这个操作生成了一个中间表示 Wh1。
Wh2 = torch.matmul(x, self.a[self.output_dim:, :])
#计算输入数据 x 和参数向量 self.a 的后半部分的矩阵乘积。这里 self.a[self.output_dim:, :] 表示选取 self.a 从 output_dim 行开始到最后,保持列不变。这个操作生成了另一个中间表示 Wh2。
e = F.leaky_relu(Wh1 + Wh2.T,negative_slope=self.alpha)
#将 Wh1 和 Wh2 的转置相加,然后通过 LeakyReLU 激活函数。LeakyReLU 激活函数允许负值有一个非零的梯度,
#由 negative_slope 参数控制,这里使用类实例的 alpha 属性作为该参数的值。这个操作生成了注意力分数 e
return e
#返回计算得到的注意力分数 e
#这个方法的目的是为注意力机制准备输入,通过将输入数据 x 与参数向量 self.a 的不同部分进行矩阵乘积,然后将结果相加并应用 LeakyReLU 激活函数,最终得到一个表示输入数据重要性的分数 e。这个分数可以用于加权输入数据,使得模型能够关注更重要的信息。这种方法是实现自注意力(self-attention)或内部注意力(intra-attention)机制的一种方式。
def forward(self,x,adj):
#用于实现前向传播过程。这个方法接受两个参数:self(指向类实例的引用),x(节点的特征矩阵),和 adj(邻接矩阵,用于表示图结构中节点之间的关系)
h = torch.matmul(x, self.weight)
#计算输入特征矩阵 x 和权重矩阵 self.weight 的矩阵乘积,得到中间表示 h。
e = self._prepare_attentional_mechanism_input(h)
#调用辅助方法 _prepare_attentional_mechanism_input 来准备注意力机制的输入,得到注意力分数 e。
zero_vec = -9e15 * torch.ones_like(e)
#创建一个与 e 形状相同、所有元素都是 -9e15 的向量。这个值非常小,接近于负无穷,用于在应用 softmax 函数之前将不相关的注意力分数置为接近零的值。
attention = torch.where(adj.to_dense()>0, e, zero_vec)
attention = F.softmax(attention, dim=1)
# attention = F.softmax(e, dim=1)
#使用 torch.where 函数根据邻接矩阵 adj 的稠密表示来选择 e 或 zero_vec。如果 adj 中的元素大于0(表示节点之间有边相连),则选择 e 中的对应值;否则,选择 zero_vec 中的值
# #attention 张量应用 softmax 函数,沿着维度1(即每个节点的注意力分数)进行归一化,使得每个节点的注意力分数之和为1。
attention = F.dropout(attention, training=self.training)
#如果模型处于训练模式(self.training 为True),则对注意力分数应用 dropout,这是一种正则化技术,用于防止过拟合。
h_pass = torch.matmul(attention, h)
#计算注意力分数矩阵 attention 和中间表示 h 的矩阵乘积,得到加权的节点特征表示 h_pass。
output_data = h_pass
#将加权的节点特征表示保存到 output_data。
output_data = F.leaky_relu(output_data,negative_slope=self.alpha)
#对 output_data 应用 LeakyReLU 激活函数。
output_data = F.normalize(output_data,p=2,dim=1)
#对 output_data 应用 L2 范数归一化,沿着维度1(即每个节点的特征向量)进行。
if self.bias is not None:
output_data = output_data + self.bias
#如果存在偏执值,则将偏执值加到output_data上
return output_data
#这个方法实现了一个注意力机制,它根据图结构中的边来加权节点特征,然后通过激活函数和归一化来生成最终的节点表示。这种注意力机制可以增强模型对图结构中重要节点的关注,从而提高图相关任务的性能。
Train_Test_Split
import pandas as pd
#pandas 是一个强大的数据分析和操作库,提供了DataFrame、Series等数据结构,非常适合处理表格数据。
import numpy as np
#numpy 是一个用于科学计算的基础库,提供了多维数组对象 ndarray 和许多用于操作这些数组的函数,对于数值计算非常重要。
import os
#它提供了许多与操作系统交互的功能,如文件路径操作、环境变量访问等。
from collections import Counter
#从 collections 模块中导入 Counter 类。Counter 是一个用于计数的字典子类,可以快速统计元素出现的次数。
from sklearn.model_selection import train_test_split
#从 sklearn.model_selection 模块中导入 train_test_split 函数。
#这个函数用于将数据集分割为训练集和测试集,是机器学习中常用的数据预处理步骤。
from utils import Network_Statistic
import argparse
parser = argparse.ArgumentParser()
#使用了 Python 的 argparse 模块来定义和解析命令行参数。argparse 是 Python 标准库的一部分,用于编写用户友好的命令行接口
parser.add_argument('--ratio', type=float, default=0.67, help='the ratio of the training set')
parser.add_argument('--num', type=int, default= 500, help='network scale')
#__num接受一个整数(type=int),默认值为 500。这个参数用于指定网络的规模
parser.add_argument('--p_val', type=float, default=0.5, help='the position of the target with degree equaling to one')
#定义了一个名为 --p_val 的命令行参数,它接受一个浮点数(type=float),默认值为 0.5。这个参数用于指定目标节点(度数为1的节点)的位置。
parser.add_argument('--data', type=str, default='hESC', help='data type')
#定义了一个名为 --data 的命令行参数,它接受一个字符串(type=str),默认值为 'hESC'。这个参数用于指定数据类型。
parser.add_argument('--net', type=str, default='Specific', help='network type')
#定义了一个名为 --net 的命令行参数,它接受一个字符串(type=str),默认值为 'Specific'。这个参数用于指定网络类型。
args = parser.parse_args()
#调用 ArgumentParser 对象的 parse_args 方法来解析命令行参数。
#这个方法会从 sys.argv 中读取命令行参数,并根据之前定义的参数进行解析。解析后的参数会被存储在一个名为 args 的命名空间对象中。
def train_val_test_set(label_file,Gene_file,TF_file,train_set_file,val_set_file,test_set_file,density,p_val=args.p_val):
#这段代码定义了一个名为 train_val_test_set 的函数,它用于从给定的标签文件、基因文件和转录因子(TF)文件中创建训练集、验证集和测试集。
#这个函数特别适用于生物信息学或网络分析中的机器学习任务,其中需要区分正样本(例如,转录因子与其目标基因的相互作用)和负样本(没有相互作用的随机选择的基因对)。
#abel_file:包含转录因子与其目标基因相互作用的标签文件的路径。
#Gene_file:包含所有基因的文件的路径。
#TF_file:包含所有转录因子的文件的路径。
#train_set_file:训练集输出文件的路径。
#val_set_file:验证集输出文件的路径。
#test_set_file:测试集输出文件的路径。
#density:用于确定测试集中负样本数量的密度参数。
#p_val:一个浮点数,默认值通过命令行参数 --p_val 指定,
#用于决定当一个转录因子只有一个目标基因时,该样本是放入训练集还是测试集中的概率。
gene_set = pd.read_csv(Gene_file, index_col=0)['index'].values
tf_set = pd.read_csv(TF_file, index_col=0)['index'].values
#这两行代码读取基因和转录因子的列表,并将其存储在 gene_set 和 tf_set 数组中。
#index_col=0 表示文件的第一列用作索引,['index'] 表示选择名为 'index' 的列(通常这是基因或转录因子的唯一标识符)
label = pd.read_csv(label_file, index_col=0)
tf = label['TF'].values
#这里读取了标签文件,并将其存储在 label DataFrame中。然后,提取了转录因子列,并将其存储在 tf 数组中。
tf_list = np.unique(tf)
pos_dict = {}
for i in tf_list:
pos_dict[i] = []
for i, j in label.values:
pos_dict[i].append(j)
#首先,使用 np.unique 函数获取所有唯一的转录因子。
#然后,创建一个空字典 pos_dict,其键是转录因子,值是一个空列表。接下来,遍历标签数据,将每个转录因子及其对应的目标基因添加到 pos_dict 中。
train_pos = {}
val_pos = {}
test_pos = {}
for k in pos_dict.keys():
if len(pos_dict[k]) <= 1:
p = np.random.uniform(0,1)
if p <= p_val:
train_pos[k] = pos_dict[k]
else:
test_pos[k] = pos_dict[k]
elif len(pos_dict[k]) == 2:
train_pos[k] = [pos_dict[k][0]]
test_pos[k] = [pos_dict[k][1]]
else:
np.random.shuffle(pos_dict[k])
train_pos[k] = pos_dict[k][:len(pos_dict[k]) * 2 // 3]
test_pos[k] = pos_dict[k][len(pos_dict[k]) * 2 // 3:]
val_pos[k] = train_pos[k][:len(train_pos[k])//5]
train_pos[k] = train_pos[k][len(train_pos[k])//5:]
#这部分代码根据转录因子的目标基因数量,将正样本分配到训练集、验证集和测试集中:
#如果一个转录因子只有一个目标基因,以 p_val 概率决定放入训练集或测试集。
#如果一个转录因子有两个目标基因,第一个放入训练集,第二个放入测试集。
#如果一个转录因子有超过两个目标基因,随机打乱目标基因列表,然后按照 2/3 和 1/3 的比例
#分配到训练集和测试集中。验证集从训练集中进一步划分,比例为 1/5。
train_neg = {}
#初始化一个空字典 train_neg,用于存储每个转录因子对应的负样本列表。
for k in train_pos.keys():
#遍历 train_pos 字典的键,这些键是转录因子。
train_neg[k] = []
#对于每个转录因子 k,在 train_neg 字典中创建一个空列表,用于存储该转录因子的负样本
for i in range(len(train_pos[k])):
#对于每个转录因子 k,遍历其对应的正样本列表 train_pos[k]。
neg = np.random.choice(gene_set)
#从所有基因的集合 gene_set 中随机选择一个基因作为潜在的负样本。
while neg == k or neg in pos_dict[k] or neg in train_neg[k]:
#检查随机选择的基因 neg 是否满足以下条件:neg == k:潜在的负样本是否与转录因子 k 相同。这是不允许的,因为负样本不能是转录因子本身。
#潜在的负样本是否已经在 pos_dict 字典中被列为转录因子 k 的正样本。这是不允许的,因为负样本不能是已知的正样本。
#潜在的负样本是否已经在 train_neg 列表中被选为该转录因子的负样本。这是不允许的,以避免重复选择同一个负样本。
neg = np.random.choice(gene_set)
#如果 neg 满足上述任何条件之一,则继续在循环中执行,再次从 gene_set 中随机选择一个新的基因。
train_neg[k].append(neg)
#一旦找到一个不满足上述条件的基因,将其添加到 train_neg[k] 列表中,作为转录因子 k 的一个负样本。
#通过这个过程,为训练集中的每个转录因子生成了一组负样本,这些负样本与转录因子没有已知的相互作用,
#也不是已经作为正样本被选中的基因。这有助于在训练机器学习模型时提供平衡的数据,使模型能够更好地区分真正的相互作用和随机的基因对。
train_pos_set = []
#初始化一个空列表,用于存储所有正样本对
train_neg_set = []
#用于存储所有的负样本对
for k in train_pos.keys():
#遍历 train_pos 字典的键,这些键代表不同的转录因子。
for j in train_pos[k]:
#对于每个转录因子 k,遍历其对应的目标基因列表 train_pos[k]。
train_pos_set.append([k, j])
#将每个转录因子 k 和其对应的目标基因 j 组成的一对添加到 train_pos_set 列表中。这个列表将包含所有的正样本对。
tran_pos_label = [1 for _ in range(len(train_pos_set))]
#创建一个标签列表 tran_pos_label,其中每个元素都是 1。
#这些标签对应于 train_pos_set 中的每个正样本对,表示它们是正样本(即转录因子和目标基因之间存在相互作用)。
#在这段代码执行完毕后,train_pos_set 将包含所有的正样本对,而 tran_pos_label 将包含与这些正样本对相对应的标签,用于训练机器学习模型时区分正负样本。
#标签 1 表示对应的样本是正样本,即转录因子和目标基因之间确实存在相互作用。
for k in train_neg.keys():
#遍历 train_neg 字典的键,这些键代表不同的转录因子。
for j in train_neg[k]:
#对于每个转录因子 k,遍历其对应的负样本列表 train_neg[k]。负样本是指随机选择的基因,这些基因不是该转录因子的目标基因。
train_neg_set.append([k, j])
#将每个转录因子 k 和其对应的负样本基因 j 组成的一对添加到 train_neg_set 列表中。这个列表将包含所有的负样本对
tran_neg_label = [0 for _ in range(len(train_neg_set))]
#创建一个标签列表 tran_neg_label,其中每个元素都是 0。这些标签对应于 train_neg_set 中的每个负样本对,表示它们是负样本(即转录因子和目标基因之间不存在相互作用)。
#在这段代码执行完毕后,train_neg_set 将包含所有的负样本对,而 tran_neg_label 将包含与这些负样本对相对应的标签,用于训练机器学习模型时区分正负样本。标签 0 表示对应的样本是负样本,即转录因子和目标基因之间不存在已知的相互作用。
train_set = train_pos_set + train_neg_set
#将包含正样本对的列表 train_pos_set 和包含负样本对的列表 train_neg_set 合并,形成完整的训练样本集 train_set。
train_label = tran_pos_label + tran_neg_label
#将包含正样本标签的列表 tran_pos_label(全部为1)和包含负样本标签的列表 tran_neg_label(全部为0)合并,形成完整的标签集 train_label。
train_sample = train_set.copy()
#复制 train_set 列表到 train_sample,用于创建带有标签的DataFrame。
for i, val in enumerate(train_sample):
val.append(train_label[i])
#遍历 train_sample 列表,enumerate 函数同时返回索引 i 和值 val。
#对于 train_sample 中的每个样本,追加对应的标签 train_label[i] 到样本列表 val 的末尾。
train = pd.DataFrame(train_sample, columns=['TF', 'Target', 'Label'])
#创建一个Pandas DataFrame train,将带有标签的样本列表 train_sample 转换为表格形式。DataFrame的列名分别设置为 'TF'、'Target' 和 'Label'。
train.to_csv(train_set_file)
#将DataFrame train 保存为CSV文件,文件路径由参数 train_set_file 指定。这样,训练集就准备好了,可以用于机器学习模型的训练
#通过以上步骤,函数将生成一个包含转录因子、目标基因和它们之间是否存在相互作用的标签的训练集,并将这个训练集保存为CSV文件,以便后续用于训练机器学习模型。
val_pos_set = []
#初始化一个空列表 val_pos_set,用于存储所有正样本对。
for k in val_pos.keys():
#遍历 val_pos 字典的键,这些键代表不同的转录因子。
for j in val_pos[k]:
#对于每个转录因子 k,遍历其对应的目标基因列表 val_pos[k]。
val_pos_set.append([k, j])
#将每个转录因子 k 和其对应的目标基因 j 组成的一对添加到 val_pos_set 列表中。这个列表将包含所有的正样本对。
val_pos_label = [1 for _ in range(len(val_pos_set))]
#创建一个标签列表 val_pos_label,其中每个元素都是 1。这些标签对应于 val_pos_set 中的每个正样本对,
#表示它们是正样本(即转录因子和目标基因之间存在相互作用
val_neg = {}
#初始化一个空字典 val_neg,用于存储每个转录因子对应的负样本列表。
for k in val_pos.keys():
#遍历 val_pos 字典的键,这些键代表不同的转录因子,这些转录因子在验证集中有正样本。
val_neg[k] = []
#对于每个转录因子 k,在 val_neg 字典中创建一个空列表,用于存储该转录因子的负样本。
for i in range(len(val_pos[k])):
#对于每个转录因子 k,遍历其对应的正样本列表 val_pos[k] 的长度。
#range(len(val_pos[k])) 生成一个从0到 val_pos[k] 列表长度减1的序列。
neg = np.random.choice(gene_set)
#从所有基因的集合 gene_set 中随机选择一个基因作为潜在的负样本。
while neg == k or neg in pos_dict[k] or neg in train_neg[k] or neg in val_neg[k]:
#neg == k:潜在的负样本是否与转录因子 k 相同。这是不允许的,因为负样本不能是转录因子本身。
#neg in pos_dict[k]:潜在的负样本是否已经在 pos_dict 字典中被列为转录因子 k 的正样本。这是不允许的,因为负样本不能是已知的正样本。
#neg in train_neg[k]:潜在的负样本是否已经在训练集的负样本列表中。这是不允许的,以避免在不同数据集中使用相同的负样本。
#neg in val_neg[k]:潜在的负样本是否已经在 val_neg 列表中被选为该转录因子的负样本。这是不允许的,以避免重复选择同一个负样本。
neg = np.random.choice(gene_set)
#如果 neg 满足上述任何条件之一,则继续在循环中执行,再次从 gene_set 中随机选择一个新的基因。
val_neg[k].append(neg)
#一旦找到一个不满足上述条件的基因,将其添加到 val_neg[k] 列表中,作为转录因子 k 的一个负样本。
#通过这个过程,为验证集中的每个转录因子生成了一组负样本,这些负样本与转录因子没有已知的相互作用,也不是已经作为正样本或训练集中的负样本被选中的基因。
#这有助于在验证机器学习模型时提供平衡的数据,使模型能够更好地评估其在未见数据上的表现。
val_neg_set = []
#初始化一个空列表 val_neg_set,这个列表将用来存储所有的负样本对。
for k in val_neg.keys():
#遍历 val_neg 字典的键,这些键是转录因子,每个转录因子都有一个与之关联的负样本列表。
for j in val_neg[k]:
#对于每个转录因子 k,遍历其对应的负样本列表 val_neg[k]。
val_neg_set.append([k,j])
#对于每个负样本,将转录因子 k 和负样本基因 j 组成的一对添加到列表 val_neg_set 中。
#这里的负样本基因 j 是从所有基因中随机选择的,且不是转录因子 k 的目标基因。
#执行完这段代码后,val_neg_set 将包含所有为验证集生成的负样本对。
#这些样本对可以用来评估机器学习模型在区分转录因子和非目标基因之间关系的能力。
#在验证过程中,模型应该能够以较高的准确率将这些负样本识别为非相互作用对。
val_neg_label = [0 for _ in range(len(val_neg_set))]
# #创建一个标签列表 val_neg_label,其中每个元素都是 0。
#这些标签对应于 val_neg_set 中的每个负样本对,表示它们是负样本(即转录因子和目标基因之间不存在相互作用)。
val_set = val_pos_set + val_neg_set
#将包含正样本对的列表 val_pos_set 和包含负样本对的列表 val_neg_set 合并,形成完整的验证样本集 val_set。
val_set_label = val_pos_label + val_neg_label
#将包含正样本标签的列表 val_pos_label(全部为1)和包含负样本标签的列表 val_neg_label(全部为0)合并,形成完整的标签集 val_set_label。
#执行完这段代码后,val_set 将包含所有的验证样本对,而 val_set_label 将包含与这些样本对相对应的标签,用于验证机器学习模型时区分正负样本。标签 1 表示对应的样本是正样本,即转录因子和目标基因之间存在相互作用;标签 0 表示对应的样本是负样本,即转录因子和目标基因之间不存在已知的相互作用。
val_set_a = np.array(val_set)
#将列表 val_set 转换成NumPy数组 val_set_a。这个数组的每一行包含一个样本,第一列是转录因子,第二列是目标基因。
val_sample = pd.DataFrame()
#创建一个空的Pandas DataFrame val_sample,用于存储验证集的样本和标签。
val_sample['TF'] = val_set_a[:,0]
#将 val_set_a 数组的第一列(转录因子)赋值给 val_sample DataFrame的新列 'TF'。
val_sample['Target'] = val_set_a[:,1]
#将 val_set_a 数组的第二列(目标基因)赋值给 val_sample DataFrame的新列 'Target'。
val_sample['Label'] = val_set_label
#将列表 val_set_label(包含所有样本的标签)赋值给 val_sample DataFrame的新列 'Label'。
val_sample.to_csv(val_set_file)
#将 val_sample DataFrame保存为CSV文件。文件路径由参数 val_set_file 指定。这样,验证集就准备好了,可以用于机器学习模型的验证。
#通过以上步骤,函数将生成一个包含转录因子、目标基因和它们之间是否存在相互作用的标签的验证集,
#并将这个验证集保存为CSV文件,以便后续用于评估机器学习模型的性能。
test_pos_set = []
#初始化一个空列表 test_pos_set,这个列表将用来存储所有的正样本对,即那些已知的转录因子与其目标基因的配对。
for k in test_pos.keys():
#遍历 test_pos 字典的键,这些键代表不同的转录因子。test_pos 字典在之前的代码中被填充,其中包含了分配给测试集的正样本。
for j in test_pos[k]:
#对于每个转录因子 k,遍历其对应的目标基因列表 test_pos[k]。这个列表包含了该转录因子的所有正样本目标基因。
test_pos_set.append([k, j])
#对于每一对转录因子 k 和目标基因 j,将它们作为一个列表 [k, j] 添加到
#test_pos_set 列表中。这样,test_pos_set 最终将包含所有测试集中的正样本对。
count = 0
#初始化一个计数器 count,用于计算测试集中所有正样本的数量。
for k in test_pos.keys():
#遍历 test_pos 字典的键,这些键代表测试集中的各个转录因子。
count += len(test_pos[k])
#对于每个转录因子 k,将其对应的正样本数量(len(test_pos[k]))加到计数器 count 上。
test_neg_num = int(count // density-count)
#计算需要生成的负样本数量。这个数量是基于正样本总数 count 和密度参数 density 计算得出的。count // density 计算在给定密度下,正样本所能占据的“槽位”,然后从这个数量中减去已有的正样本数量,得到需要的负样本数量。结果转换为整数,因为样本数量必须是整数。
test_neg = {}
#初始化一个空字典 test_neg,用于存储每个转录因子的负样本列表。
for k in tf_set:
#遍历 tf_set 集合中的所有转录因子。tf_set 包含了所有可能的转录因子。
test_neg[k] = []
#对于每个转录因子 k,在 test_neg 字典中创建一个空列表,用于存储该转录因子的负样本。
test_neg_set = []
#初始化一个空列表 test_neg_set,用于存储生成的负样本对。
for i in range(test_neg_num):
#循环 test_neg_num 次,test_neg_num 是之前计算出的需要生成的负样本数量。
t1 = np.random.choice(tf_set)
#从 tf_set 集合中随机选择一个转录因子 t1。tf_set 包含了所有可能的转录因子。
t2 = np.random.choice(gene_set)
#从 gene_set 集合中随机选择一个基因 t2。gene_set 包含了所有可能的基因。
while t1 == t2 or [t1, t2] in train_set or [t1, t2] in test_pos_set or [t1, t2] in val_set or [t1,t2] in test_neg_set:
#t1 == t2:确保转录因子 t1 和基因 t2 不相同。
#[t1, t2] in train_set:确保样本对 (t1, t2) 不在训练集中。
#[t1, t2] in test_pos_set:确保样本对 (t1, t2) 不在测试集的正样本中。
#[t1, t2] in val_set:确保样本对 (t1, t2) 不在验证集中。
#[t1, t2] in test_neg_set:确保样本对 (t1, t2) 不在已生成的测试集负样本中。
t2 = np.random.choice(gene_set)
#如果当前选择的 t2 不满足条件,则重新从 gene_set 中选择一个新的基因。
test_neg_set.append([t1,t2])
#一旦找到满足所有条件的 t1 和 t2,将这对样本添加到 test_neg_set 列表中。
test_pos_label = [1 for _ in range(len(test_pos_set))]
#创建一个标签列表 test_pos_label,其中每个元素都是 1。
#这些标签对应于 test_pos_set 中的每个正样本对,表示它们是正样本(即转录因子和目标基因之间存在相互作用)。
test_neg_label = [0 for _ in range(len(test_neg_set))]
#创建一个标签列表 test_neg_label,其中每个元素都是 0。
#这些标签对应于 test_neg_set 中的每个负样本对,表示它们是负样本(即转录因子和目标基因之间不存在相互作用)
test_set = test_pos_set + test_neg_set
#将包含正样本对的列表 test_pos_set 和包含负样本对的列表 test_neg_set 合并,
#形成完整的测试样本集 test_set。这个集合包含了所有用于测试的样本对,无论它们是正样本还是负样本。
test_label = test_pos_label + test_neg_label
#将包含正样本标签的列表 test_pos_label(全部为1)和包含负样本标签的列表 test_neg_label(全部为0)合并,
#形成完整的标签集 test_label。这个集合包含了所有测试样本对的标签,用于指示每个样本对是正样本还是负样本。
for i, val in enumerate(test_set):
val.append(test_label[i])
#对于 test_set 中的每个样本对,追加对应的标签 test_label[i] 到样本对 val 的末尾。
#这样,每个样本对都会扩展为包含三个元素的列表:转录因子、目标基因和标签。
test_sample = pd.DataFrame(test_set, columns=['TF', 'Target', 'Label'])
#创建一个Pandas DataFrame test_sample。这个DataFrame由列表 test_set 中的数据构成,
#其中每个元素都是一个包含转录因子、目标基因和标签的列表。
#columns 参数指定了DataFrame的列名,分别是 'TF'、'Target' 和 'Label',分别对应转录因子、目标基因和它们之间是否存在相互作用的标签。
test_sample.to_csv(test_set_file)
#将 test_sample DataFrame保存为CSV文件。文件路径由参数 test_set_file 指定。这样,测试集就准备好了,可以用于机器学习模型的测试。
def Hard_Negative_Specific_train_test_val(label_file, Gene_file, TF_file, train_set_file,val_set_file,test_set_file,
ratio=args.ratio, p_val=args.p_val):
#它用于创建训练集、验证集和测试集,特别关注于“硬负样本”(hard negative samples)。
#这些是模型可能会错误分类为正样本的负样本,因为它们在某些特征上与正样本相似
#包含转录因子(TF)和目标基因之间相互作用的标签文件路径。
#Gene_file:包含所有基因的文件路径。
#TF_file:包含所有转录因子的文件路径。
#train_set_file:训练集输出文件的路径。
#val_set_file:验证集输出文件的路径。
#test_set_file:测试集输出文件的路径。
#ratio:用于控制训练集、验证集和测试集划分比例的参数,它有一个默认值,通过命令行参数 --ratio 指定。
#p_val:用于决定当一个转录因子只有一个目标基因时,该样本是放入训练集还是测试集中的概率,它有一个默认值,通过命令行参数 --p_val 指定。
label = pd.read_csv(label_file, index_col=0)
#使用 pandas 库读取标签文件,并将第一列作为索引。
gene_set = pd.read_csv(Gene_file, index_col=0)['index'].values
#读取基因文件,并将名为 'index' 的列(通常包含基因的唯一标识符)转换为数组 gene_set。
tf_set = pd.read_csv(TF_file, index_col=0)['index'].values
#读取转录因子文件,并将名为 'index' 的列转换为数组 tf_set。
tf = label['TF'].values
#从标签文件中提取转录因子列,并将其转换为数组 tf。
tf_list = np.unique(tf)
#使用 numpy 库的 unique 函数获取数组 tf 中的唯一值,这些唯一值代表所有不同的转录因子,并存储在数组 tf_list 中
pos_dict = {}
#初始化一个空字典 pos_dict,这个字典将用于存储正样本数据,即转录因子和它们的目标基因之间的相互作用。
for i in tf_list:
pos_dict[i] = []
#遍历包含所有唯一转录因子的数组 tf_list。
#对于每个转录因子 i,在字典 pos_dict 中创建一个空列表,这个列表将用来存储与该转录因子相互作用的目标基因。
for i, j in label.values:
#for i, j in label.values 会同时遍历转录因子(i)和对应的目标基因(j)。
pos_dict[i].append(j)
#对于每一对转录因子和目标基因,将目标基因 j 添加到对应转录因子 i 的列表中。
#这样,pos_dict 字典将包含所有转录因子及其对应的目标基因列表。
neg_dict = {}
#初始化一个空字典 neg_dict,这个字典将用于存储每个转录因子对应的负样本列表。
for i in tf_set:
#遍历包含所有转录因子的数组 tf_set。
neg_dict[i] = []
#对于每个转录因子 i,在字典 neg_dict 中创建一个空列表,
#这个列表将用来存储与该转录因子不相互作用的基因(负样本)。
for i in tf_set:
if i in pos_dict.keys():
#检查当前转录因子 i 是否存在于正样本字典 pos_dict 的键中。
pos_item = pos_dict[i]
#如果存在,获取与转录因子 i 相关的正样本列表 pos_item。
pos_item.append(i)
#将转录因子 i 本身添加到正样本列表 pos_item 中。这一步可能是为了确保转录因子本身不被错误地视为负样本。
neg_item = np.setdiff1d(gene_set, pos_item)
#使用 numpy 库的 setdiff1d 函数计算 gene_set 和 pos_item 之间的差集,得到与转录因子 i 不相关的基因列表 neg_item。
neg_dict[i].extend(neg_item)
#将负样本列表 neg_item 添加到 neg_dict 字典中对应转录因子 i 的列表中。
pos_dict[i] = np.setdiff1d(pos_dict[i], i)
#从 pos_dict 字典中对应转录因子 i 的正样本列表中移除转录因子 i 本身。
else:
neg_item = np.setdiff1d(gene_set, i)
#计算 gene_set 和只包含转录因子 i 的列表之间的差集,得到与转录因子 i 不相关的基因列表 neg_item。
neg_dict[i].extend(neg_item)
#将负样本列表 neg_item 添加到 neg_dict 字典中对应转录因子 i 的列表中。
train_pos = {}
val_pos = {}
test_pos = {}
#train_pos:用于存储分配给训练集的正样本。
#val_pos:用于存储分配给验证集的正样本。
#test_pos:用于存储分配给测试集的正样本
for k in pos_dict.keys():
#遍历 pos_dict 字典的键,这些键是转录因子,值是对应的目标基因列表。
if len(pos_dict[k]) ==1:
#如果转录因子 k 只有一个正样本,生成一个随机数 p。
p = np.random.uniform(0,1)
if p <= p_val:
train_pos[k] = pos_dict[k]
#如果随机数 p 小于或等于 p_val,则将该正样本分配给训练集 train_pos[k]
else:
test_pos[k] = pos_dict[k]
#否则,将该正样本分配给测试集 test_pos[k]。
elif len(pos_dict[k]) ==2:
np.random.shuffle(pos_dict[k])
#如果转录因子 k 有两个正样本,随机打乱这两个样本。
train_pos[k] = [pos_dict[k][0]]
test_pos[k] = [pos_dict[k][1]]
#将第一个样本分配给训练集。将第二个样本分配给测试集。
else:#如果转录因子 k 有超过两个正样本,随机打乱这些样本。
np.random.shuffle(pos_dict[k])
train_pos[k] = pos_dict[k][:int(len(pos_dict[k])*ratio)]
#将样本列表的前 ratio 比例的部分分配给训练集。
val_pos[k] = pos_dict[k][int(len(pos_dict[k])*ratio):int(len(pos_dict[k])*(ratio+0.1))]
#将样本列表的接下来的 0.1 比例的部分分配给验证集。
test_pos[k] = pos_dict[k][int(len(pos_dict[k])*(ratio+0.1)):]
#将剩余的样本分配给测试集
train_neg = {}
val_neg = {}
test_neg = {}
#train_neg:用于存储分配给训练集的负样本。
#val_neg:用于存储分配给验证集的负样本。
#test_neg:用于存储分配给测试集的负样本。
for k in pos_dict.keys():
#遍历 pos_dict 字典的键,这些键是转录因子。
#这里使用 pos_dict 的键是因为负样本的分配应该与正样本相对应,确保每个转录因子都有相应的正负样本。
neg_num = len(neg_dict[k])
#对于每个转录因子 k,计算其负样本的数量,并存储在变量 neg_num 中。
np.random.shuffle(neg_dict[k])
#随机打乱转录因子 k 对应的负样本列表,以确保样本的随机分配。
train_neg[k] = neg_dict[k][:int(neg_num*ratio)]
#根据给定的比例 ratio,将负样本列表的前 ratio 部分分配给训练集。int(neg_num*ratio) 计算出应该分配给训练集的负样本数量。
val_neg[k] = neg_dict[k][int(neg_num*ratio):int(neg_num*(0.1+ratio))]
#将负样本列表的接下来的 0.1 比例的部分(在 ratio 之后)分配给验证集。这里 0.1 是额外的验证集比例。
test_neg[k] = neg_dict[k][int(neg_num*(0.1+ratio)):]
#将剩余的负样本分配给测试集。这意味着测试集将包含从 int(neg_num*(0.1+ratio)) 到最后的所有负样本。
train_pos_set = []
#初始化一个空列表 train_pos_set,这个列表将用来存储训练集中所有的正样本对。
for k in train_pos.keys():
#遍历 train_pos 字典的键,这些键代表分配给训练集的转录因子。
for val in train_pos[k]:
#对于每个转录因子 k,遍历其对应的正样本列表 train_pos[k],这个列表包含了与该转录因子相互作用的目标基因。
train_pos_set.append([k,val])
#对于每一对转录因子 k 和目标基因 val,将它们作为一个列表 [k, val] 添加到 train_pos_set 列表中。这样,train_pos_set 最终将包含所有训练集中的正样本对。
train_neg_set = []
#初始化一个空列表 train_neg_set,这个列表将用来存储训练集中所有的负样本对。
for k in train_neg.keys():
#遍历 train_neg 字典的键,这些键代表分配给训练集的转录因子。
for val in train_neg[k]:
#对于每个转录因子 k,遍历其对应的负样本列表 train_neg[k],这个列表包含了不是该转录因子目标基因的随机选择的基因。
train_neg_set.append([k,val])
#对于每一对转录因子 k 和负样本基因 val,将它们作为一个列表 [k, val] 添加到 train_neg_set 列表中。
#这样,train_neg_set 最终将包含所有训练集中的负样本对。
train_set = train_pos_set + train_neg_set
#将包含正样本对的列表 train_pos_set 和包含负样本对的列表 train_neg_set 合并,
#形成完整的训练样本集 train_set。这个集合包含了所有用于训练的样本对,无论它们是正样本还是负样本。
train_label = [1 for _ in range(len(train_pos_set))] + [0 for _ in range(len(train_neg_set))]
#[1 for _ in range(len(train_pos_set))] 生成一个由 1 组成的列表,每个 1 对应一个正样本,表示存在相互作用。
#[0 for _ in range(len(train_neg_set))] 生成一个由 0 组成的列表,每个 0 对应一个负样本,表示不存在相互作用。
#这两个列表合并起来,为 train_set 中的每个样本对提供正确的标签。
train_sample = np.array(train_set)
#将列表 train_set 转换成NumPy数组 train_sample。这个数组的每一行包含一个样本对,其中第一列是转录因子,第二列是目标基因。
train = pd.DataFrame()
#创建一个空的Pandas DataFrame train,用于存储训练集的样本和标签。
train['TF'] = train_sample[:, 0]
#将 train_sample 数组的第一列(转录因子)赋值给 train DataFrame的新列 'TF'。
train['Target'] = train_sample[:, 1]
#将 train_sample 数组的第二列(目标基因)赋值给 train DataFrame的新列 'Target'。
train['Label'] = train_label
#将列表 train_label(包含所有样本的标签)赋值给 train DataFrame的新列 'Label'。
train.to_csv(train_set_file)
#将 train DataFrame保存为CSV文件。文件路径由参数 train_set_file 指定。这样,训练集就准备好了,可以用于机器学习模型的训练。
val_pos_set = []
#初始化一个空列表 val_pos_set,这个列表将用来存储验证集中所有的正样本对。
for k in val_pos.keys():
#遍历 val_pos 字典的键,这些键代表分配给验证集的转录因子。
for val in val_pos[k]:
#对于每个转录因子 k,遍历其对应的正样本列表 val_pos[k],这个列表包含了与该转录因子相互作用的目标基因。
val_pos_set.append([k,val])
#对于每一对转录因子 k 和目标基因 val,将它们作为一个列表 [k, val] 添加到 val_pos_set 列表中。这样,val_pos_set 最终将包含所有验证集中的正样本对。
val_neg_set = []
#初始化一个空列表 val_neg_set,这个列表将用来存储验证集中所有的负样本对。
for k in val_neg.keys():
#遍历 val_neg 字典的键,这些键代表分配给验证集的转录因子。
for val in val_neg[k]:
#对于每个转录因子 k,遍历其对应的负样本列表 val_neg[k],这个列表包含了不是该转录因子目标基因的随机选择的基因。
val_neg_set.append([k,val])
#对于每一对转录因子 k 和负样本基因 val,将它们作为一个列表 [k, val] 添加到 val_neg_set 列表中。这样,val_neg_set 最终将包含所有验证集中的负样本对。
val_set = val_pos_set + val_neg_set
#将包含正样本对的列表 val_pos_set 和包含负样本对的列表 val_neg_set 合并,
#形成完整的验证样本集 val_set。这个集合包含了所有用于验证的样本对,无论它们是正样本还是负样本。
val_label = [1 for _ in range(len(val_pos_set))] + [0 for _ in range(len(val_neg_set))]
#[1 for _ in range(len(val_pos_set))] 生成一个由 1 组成的列表,每个 1 对应一个正样本,表示存在相互作用。
#[0 for _ in range(len(val_neg_set))] 生成一个由 0 组成的列表,每个 0 对应一个负样本,表示不存在相互作用。
#这两个列表合并起来,为 val_set 中的每个样本对提供正确的标签。
val_sample = np.array(val_set)
#将列表 val_set 转换成NumPy数组 val_sample。这个数组的每一行包含一个样本对,其中第一列是转录因子,第二列是目标基因。
val = pd.DataFrame()
#创建一个空的Pandas DataFrame val,用于存储验证集的样本和标签。
val['TF'] = val_sample[:, 0]
#将 val_sample 数组的第一列(转录因子)赋值给 val DataFrame的新列 'TF'。
val['Target'] = val_sample[:, 1]
#将 val_sample 数组的第二列(目标基因)赋值给 val DataFrame的新列 'Target'。
val['Label'] = val_label
#将列表 val_label(包含所有样本的标签)赋值给 val DataFrame的新列 'Label'。
val.to_csv(val_set_file)
#将 val DataFrame保存为CSV文件。文件路径由参数 val_set_file 指定。这样,验证集就准备好了,可以用于机器学习模型的验证。
test_pos_set = []
#初始化一个空列表 test_pos_set,这个列表将用来存储测试集中所有的正样本对。
for k in test_pos.keys():
#遍历 test_pos 字典的键,这些键代表分配给测试集的转录因子。
for j in test_pos[k]:
#对于每个转录因子 k,遍历其对应的正样本列表 test_pos[k],这个列表包含了与该转录因子相互作用的目标基因。
test_pos_set.append([k,j])
#对于每一对转录因子 k 和目标基因 j,将它们作为一个列表 [k, j] 添加到 test_pos_set 列表中。
#这样,test_pos_set 最终将包含所有测试集中的正样本对。
test_neg_set = []
#初始化一个空列表 test_neg_set,这个列表将用来存储测试集中所有的负样本对。
for k in test_neg.keys():
#遍历 test_neg 字典的键,这些键代表分配给测试集的转录因子。
for j in test_neg[k]:
#对于每个转录因子 k,遍历其对应的负样本列表 test_neg[k],这个列表包含了不是该转录因子目标基因的随机选择的基因。
test_neg_set.append([k,j])
#对于每一对转录因子 k 和负样本基因 j,将它们作为一个列表 [k, j] 添加到 test_neg_set 列表中。
#这样,test_neg_set 最终将包含所有测试集中的负样本对。
test_set = test_pos_set +test_neg_set
#将包含正样本对的列表 test_pos_set 和包含负样本对的列表 test_neg_set 合并,
#形成完整的测试样本集 test_set。这个集合包含了所有用于测试的样本对,无论它们是正样本还是负样本。
test_label = [1 for _ in range(len(test_pos_set))] + [0 for _ in range(len(test_neg_set))]
#[1 for _ in range(len(test_pos_set))] 生成一个由 1 组成的列表,每个 1 对应一个正样本,表示存在相互作用。
#这两个列表合并起来,为 test_set 中的每个样本对提供正确的标签。
#test_set 将包含所有的测试样本对,而 test_label 将包含与这些样本对相对应的标签,用于测试机器学习模型时区分正负样本。
#标签 1 表示对应的样本是正样本,即转录因子和目标基因之间存在相互作用;标签 0 表示对应的样本是负样本,即转录因子和目标基因之间不存在已知的相互作用。
#这种标签化是监督学习中的关键步骤,它允许模型在测试过程中评估其在未见数据上的性能。
test_sample = np.array(test_set)
#将列表 test_set 转换成NumPy数组 test_sample。这个数组的每一行包含一个样本对,其中第一列是转录因子,第二列是目标基因。
test = pd.DataFrame()
#创建一个空的Pandas DataFrame test,用于存储测试集的样本和标签。
test['TF'] = test_sample[:,0]
#将 test_sample 数组的第一列(转录因子)赋值给 test DataFrame的新列 'TF'。
test['Target'] = test_sample[:,1]
test['Label'] = test_label
test.to_csv(test_set_file)
#将 test DataFrame保存为CSV文件。文件路径由参数 test_set_file 指定。这样,测试集就准备好了,可以用于机器学习模型的测试。
if __name__ == '__main__':
#这是一个常用的Python模式,用于确定代码是否在作为脚本直接运行时执行,而不是在作为模块导入时执行
data_type = args.data
net_type = args.net
#从命令行参数中获取 data_type 和 net_type 的值,分别代表数据类型和网络类型。
density = Network_Statistic(data_type=data_type, net_scale=args.num, net_type=net_type)
#使用 Network_Statistic 类(可能是一个自定义类)计算网络统计信息,如密度。这个密度值可能用于后续的数据集划分。
TF2file = os.getcwd() + '/' + net_type + ' Dataset/' + data_type + '/TFs+' + str(args.num) + '/TF.csv'
Gene2file = os.getcwd() + '/' + net_type + ' Dataset/' + data_type + '/TFs+' + str(args.num) + '/Target.csv'
label_file = os.getcwd() + '/' + net_type + ' Dataset/' + data_type + '/TFs+' + str(args.num) + '/Label.csv'
train_set_file = os.getcwd() + '/.../' + net_type + '/' + data_type + ' ' + str(args.num) + '/Train_set.csv'
test_set_file = os.getcwd() + '/.../' + net_type + '/' + data_type + ' ' + str(args.num) + '/Test_set.csv'
val_set_file = os.getcwd() + '/.../' + net_type + '/' + data_type + ' ' + str(args.num) + '/Validation_set.csv'
#构建转录因子文件、目标基因文件和标签文件的路径,以及训练集、测试集和验证集文件的路径。这些路径基于当前工作目录、网络类型、数据类型和网络规模。
path = os.getcwd() + '/.../' + net_type + '/' + data_type + ' ' + str(args.num)
#构建输出路径,包括网络类型、数据类型和网络规模。
if not os.path.exists(path):
os.makedirs(path)
#检查输出路径是否存在,如果不存在,则创建它。
if net_type == 'Specific':
Hard_Negative_Specific_train_test_val(label_file, Gene2file, TF2file, train_set_file, val_set_file,
test_set_file)
#如果 net_type 是 'Specific',则调用 Hard_Negative_Specific_train_test_val 函数来生成训练集、验证集和测试集。
else:
train_val_test_set(label_file, Gene2file, TF2file, train_set_file, val_set_file, test_set_file, density)
#否则,调用 train_val_test_set 函数来生成数据集,并且可能使用计算出的 density 值。
首先我们要知道Lable是一个关于TF和Target的文件,Target文件是一个关于Gene和index的文件,TF文件是一个关于TF和index的文件(主要这里的index与系统标注的序号是不一致的)
然后来走这个Train_Test_Split文件,第一个赋值的时候是数据类型是hESC,网络类型是Specific,(这个是如果你不选,系统自己改默认的),这个density是根据数据类型,网络尺寸,网络类型来做的,第一次得到的结果是0.164.,第一次走会走下面这个train_val_test_set(),接下来进入这段代码来看,第一句gene_set得到的是Gene 文件的index的列值(得到的是从1到909的一个列表,与系统生成的序列号是一致的),tf_set得到的是index的列值(从0到***的一个列值,注意这里与系统生成的序列号不是一致的),label会得到一个类似于TF和Target的关系的表格,主要这里是以第一列为索引,所以要查找的时候,找某一个TF会得到所有关于它的Targe,tf_list就是tf去重之后得到的一个列表,首先创建一个空字典,遍历tf_list给每一个i都创建一个对应的空列表,运行下面这个for,i对应label中的Gene列,j对应的是Targe列,运行完之后得到的pos_dict关键字对应的是label中的TF,而关键字的值对应的是label中值所对应的target,接下来创建三个空字典,遍历pos_dict中的关键字,如果这个TF所调控的Target的数量是没有边或者只有一个边的时候,就按照p_val的概率看是分到训练集中还是测试集中,如果label所对应的target的边有两个,则一个分到训练集,一个分到测试集中,若是边的数量比较多,前三分之二分到训练集,后三分之一分到测试集,验证集则在进行一个细分,训练集的前五分之一分到验证集,剩下的依然是留在训练集中,形式大概是如此,验证集和测试集大概也是如此
train _pos:{0: [1127, 755, 1118, 1143, 732, 187, 1221, 11], 2: [531, 328, 781, 1062, 560, 533]
遍历训练集中关键字,为每一个关键字创建一个空列表,neg从gene_set中随机选择一个数据, 检查随机选择的基因 neg 是否满足以下条件:neg == k:潜在的负样本是否与转录因子 k 相同。这是不允许的,因为负样本不能是转录因子本身。潜在的负样本是否已经在 pos_dict 字典中被列为转录因子 k 的正样本。这是不允许的,因为负样本不能是已知的正样本。潜在的负样本是否已经在 train_neg 列表中被选为该转录因子的负样本。这是不允许的,以避免重复选择同一个负样本。如果找不到就一直找,将找到的neg放到构建的负样本集合中,创建两个空列表,遍历正训练集中的关键字,j再来遍历正训练集中关键字对应的关键值,将这个关键字与所对应的关键值放到train_pos_set列表中,得到的结果大概是[[0, 1127], [0, 755], [0, 1118], [0, 1143], [0, 732], [0, 187]]这样一个东西,创建一个标签列表 tran_pos_label,其中每个元素都是 1,同样的,创建一个标签列表 train_neg_label,其中每个元素都是 0,然后将训练集和测试集放在一起,将训练标签和测试标签放在一起,将值复制到train_sample中,遍历 train_sample 列表,enumerate 函数同时返回索引 i 和值 val,在每一个列值val后面添加一个标签,然后将生成的这个训练集写到对应的训练集csv中,接下来是验证集,同样的道理,先看验证集中的关键字,遍历每一个关键字对应的值,将关键字对应的关键值写入到val_pos_set中,生成标签,下面创建一个负样本的空字典,遍历正验证集中的关键字,创建一个空列表,对于每一个正样本的关键字的,选择同样数量的负样本,先从gene_set列表中随机选择一个,选择的这个Gene不能等于调控因子本身,不能在已知正样本中,不能已经在验证负样本中,不能在训练负样本中,这样是为了确保随机性,将验证集中不存在调控关系的转录因子和调控基因添加的val_neg_set集合中并且赋予一个标签,验证集中正负样本集合与正负样本标签合并,然后将其转化为array格式,将其写入到验证集对应的位置,最后就是测试集的构造,遍历正测试集中的关键字,遍历每一个关键字对应的关键值,将关键字与关键值都放入到test_pos_set这个集合中,首先统计出正测试集中边的数量,计算出测试集中负样本应该构建的数量, t1 = t2:确保转录因子 t1 和基因 t2 不相同。[t1, t2] in train_set:确保样本对 (t1, t2) 不在训练集中。[t1, t2] in test_pos_set:确保样本对 (t1, t2) 不在测试集的正样本中。[t1, t2] in val_set:确保样本对 (t1, t2) 不在验证集中。[t1, t2] in test_neg_set:确保样本对 (t1, t2) 不在已生成的测试集负样本中。
接下来看Hard_Negative_Specific这个函数,第一句话来读取读取label_file文件,lable得到的是含有TF和Target两列的一种数据类型,gene_set得到的是Gene_file的index这一列的值,tf_set得到的是TF_file这一列的值,tf是从label中得到TF这一列的值,然后下一步tf_list是去重结果,定义一个空字典,遍历lable中每一个不重复的TF的值,为每一个不重复的值创建一个空列表,将每一个空列表其所对应的调控的基因添加,得到类似于这样的结果,pos_dict:{43: [825, 831, 641, 104, 244]},接下来构建负样本字典,为每一个tf_set都构造一个空列表,检查当前转录因子 i 是否存在于正样本字典 pos_dict 的键中,若是存在,将与转录因子i调控的基因赋值给pos_item,将转录因子 i 本身添加到正样本列表 pos_item 中。这一步可能是为了确保转录因子本身不被错误地视为负样本。使用 numpy 库的 setdiff1d 函数计算 gene_set 和 pos_item 之间的差集,得到与转录因子 i 不相关的基因列表 neg_item。将负样本列表 neg_item 添加到 neg_dict 字典中对应转录因子 i 的列表中。pos_dict 字典中对应转录因子 i 的正样本列表中移除转录因子 i 本身。若是不存在,直接将其减去本身之后作为转了因子i的负样本,所以本身即不作为正样本,也不作为负样本,创建三个空字典,遍历正样本空间,若是其中节点的边的数量是为1,则根据概率分到训练集中或者测试集中,若是节点的调控关系是为2,则一个分到训练集中,一个分到测试集中,若是比较多则按比例分配训练,验证,测试,将正样本与负样本合并成一个训练集,生成对应数量的标签,将训练集以及标签写入到CSV文件中,
1,也就是说训练集和验证集中存在调控关系的与不存在调控关系的边的数量是一致的,但是在测试集中并不一致,遍历调控因子那个表的index列(在代码141行),为每一个k创建一个空列表,随机的从tf文件的index列中选择一个值,从gene文件中随机选择一个值,同样的道理,将正负训练集合并在一起,将正负测试集合并在一起,在将标签合并到训练集中,将其写入到测试集所对应的位置上,
2,在Hard_Negative_Specific_train_test_val是为每一个TF文件中出现的转录因子都创建一个空列表,在train_val_test_set构建是构建与pos_dict同样数量的节点并且每一个节点中边的数量也是一致的(或者说构建的是与lable中TF的数量一致的空列表),与正样本的区别在于就是不存在调控关系而已,
3,选择训练集,验证集,测试集时,Hard_Negative_Specific_train_test_val,对于每一个节点所连接的边,都要先进行打乱,再安比例进行选择,并且这个比例与之前有所不同,之前是训练集三分之2,测试集三分之一,验证集是验证集中的五分之一,这里训练集0.67,验证集0.67到0.77之间,剩下的作为测试集,
3,构建训练集,测试集,验证集负样本的策略不同,这里也是按照训练集0.67,验证集0.67到0.77之间,剩下的作为测试集来构建的,这里相较于train_val_test_set负样本每一个集的负样本数量都是变多的,
debug
首先设置一些参数,用于后续模型的训练和测试==(main20~32),为了保证实验的可重复性,这里设置了随机种子,这是试验标准化的一部分,确保每次运行的结果的一致性(main36 ~ 40),exp_file(这个文件包含了单细胞RNA测序数据,即不同细胞中基因的表达水平) tf_file(指向包含转录因子的csv文件) target_file:(包含了目标基因信息的CSV文件) train_file(包含训练集数据的CSV文件)val_file(指向包含验证数据集的CSV文件)。指定这几个文件的路径,然后是指定tf_embed_path和target_embed_pathl路径,确保在模型训练完成后,能够将重要的中间结果(嵌入向量)保存下来,以便于后续的研究和应用(main57~66)代码加载了基因表达矩阵的数据并使用load_data类进行预处理,再使用exp_data方法将数据标准化并且将其转化为float32类型,然后将处理后的数据转化为pytorch张量,以适配PyTorch模型的输入要求,(main 72~ 78,utils 7395)==根据GPU是否可用,将数据和模型移动到CPU或者GPU上,以加速计算==(main8183)数据集和邻接矩阵准备,使用scRNADataset类来创建数据集,并生成邻接矩阵,表达基因之间的潜在相互作用,最后将稀疏矩阵格式转化为Pytorch稀疏张量,将训练数据集和验证数据都转化为张量,以便在PyTorch模型中使用(main88~94,utils 4269,98104),初始化GeneLink模型,将模型或者数据移动到CPU或者GPU上(main98114)==,使用adam优化器来跟新模型权重,StepLR学习率调度器用于在训练过程中调整学习率==(main117118),在每一个epoch中,使用 DataLoader 从 train_load 数据集中加载训练数据。batch_size 和 shuffle 参数分别控制每个批次的大小和数据是否需要打乱,调整模型为训练模式,清零模型梯度为新一列的计算做准备,并且判断是否是进行因果推断,data_feature基因表达数据,提供了每个基因在不同细胞中的表达水平,adj邻接矩阵,提供了基因之间的连接信息,特定的训练样本,这些样本的特征将被用来计算出当前批次数据的预测结果pred,tensor([[0.2726], [0.2697], [0.2734], … [0.3429],[0.3298], [0.2635]]),计算预测概率pred和真实标签train_y之间的二元交叉熵损失,进行反向传播和参数更新,将当前批次的损失累计到running_loss中(main127~158)== 接下来是评估模式,依然是传入data_feature,adj,不过评估模式传入的是验证数据集,得到类似于上述的pred不过这里命名为score,根据是否进行因果推断来处理score,将模式对验证集的预测,验证集真正的标签,是否进行因果推断传入Evaluation方法,这个函数的主要作用是计算模型的性能指标,包括AUC,和AUPR,AUPR_norm, 打印这一轮的总损失 AUC,AUPR,将模型的参数保存到指定路径,设置当前为评估模式,获的转录因子和目标基因的嵌入表示,将转录因子和目标嵌入,目标基因的路径,传入这个embed2file函数,这个函数的作用是并将这些数据写入到指定的文件中==(main4566,161182,utiles115~135)==

















 943
943

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









