







最近看到读者留言说在差异表达分析导入矩阵是提醒row name重复,现在就这一问题解释原因和最简单的解决方案。
原因:探针和基因是多对一的关系,比如A和B都可能是指向基因AB。在一般的基因芯片的表达矩阵中,用探针表示的表达矩阵不存在行名重复问题。但是如果先注释成gene symbol,则可能不同行的探针注释成同一个gene symbol。这个时候如果还是用转换后的矩阵进行差异分析,在导入R的时候就会提醒row name充分,这是由于R的规则将行名视为唯一标识符,如果由两个行具有相同的名称,在使用行名取数据的时候,R就不知道需要的是哪一行。
解决方案:通常情况下将不同探针获得的gene symbol按照一定规则合并成一行即可。可以使平均数、中位数、最大值、最小值等,根据自己的需求决定。这个操作可以在R中完成,但是需要一定的编程基础。下面以求平均数为例演示如何使用Excel合并相同的行。
1. 这是一个带有重复行名的表达矩阵,只有一个样本。
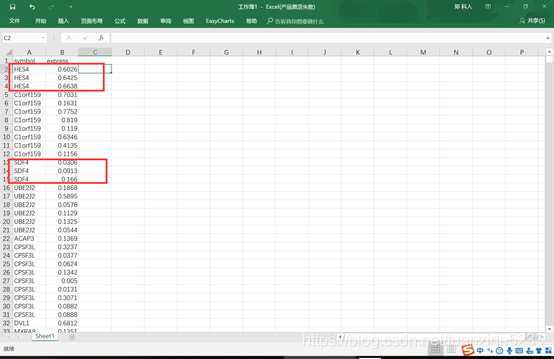
2.新建一个sheet并点击左上角的透视表
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









