






 本文介绍了如何通过WGDI工具进行共线性分析,利用Ks点阵图和频率分布图探索物种历史中的加倍事件。通过配置文件调整,过滤关键参数如显著性、串联基因和Ks范围,以获得更可靠的数据可视化结果。并与wgd工具的分析进行对比,强调WGDI在处理Ks峰值上的优势。
本文介绍了如何通过WGDI工具进行共线性分析,利用Ks点阵图和频率分布图探索物种历史中的加倍事件。通过配置文件调整,过滤关键参数如显著性、串联基因和Ks范围,以获得更可靠的数据可视化结果。并与wgd工具的分析进行对比,强调WGDI在处理Ks峰值上的优势。
Ks可视化
我们在上一篇如何用WGDI进行共线性分析(一)得到共线性分析结果和Ks值输出结果的整合表格文件后,我们就可以绘制Ks点阵图和Ks频率分布图对共线性区的Ks进行探索性分析,从而确定可能的Ks峰值,用于后续分析。
Ks点阵图
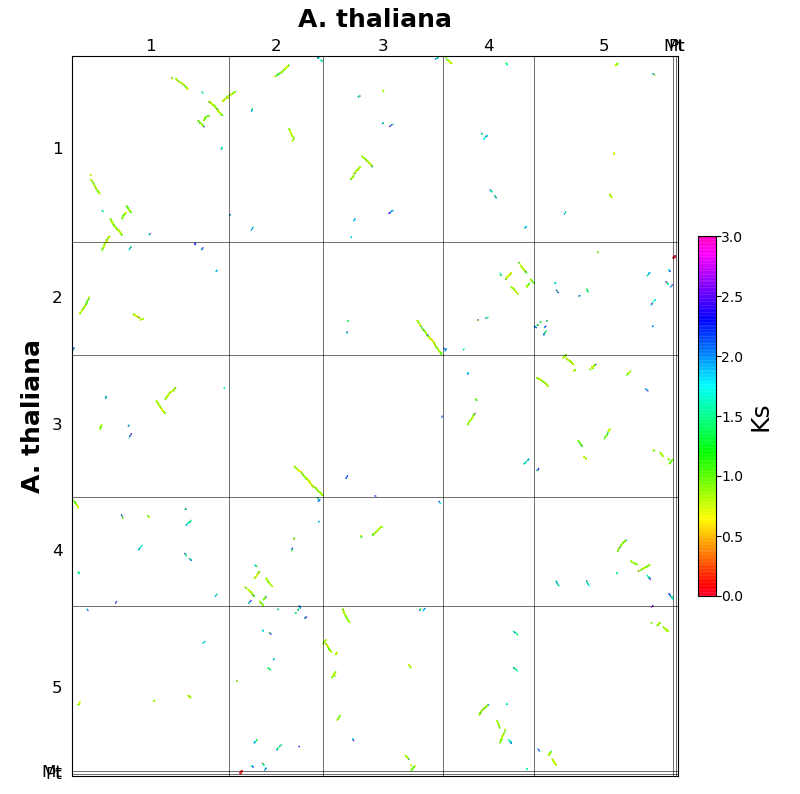
最初绘制的点图信息量很大,基本上涵盖了历史上发生的加倍事件所产生的共线性区。我们能大致的判断片段是否存在复制以及复制了多少次,至于这些片段是否来自于同一次加倍事件则不太好确定。借助于Ks信息 (Ks值可以反应一定尺度内的演化时间),我们就可以较为容易地根据点阵图上共线性区域的颜色来区分多倍化事件。
首先,创建配置文件(这次是 -bk模块,BlockKs)
wgdi -bk \? >> ath.conf然后,修改配置文件
[blockks]
lens1 = ath.len
lens2 = ath.len
genome1_name = A. thaliana
genome2_name = A. thaliana
blockinfo = ath_block_information.csv
pvalue = 0.05
tandem = true
tandem_length = 200
markersize = 1
area = 0,3
block_length = 5
figsize = 8,8
savefig = ath.ks.dotplot.pdfblockinfo 是前面共线性分析和Ks分析的整合结果,是绘图的基础。根据下面几个标准进行过滤
- pvalue: 共线性区的显著性, 对应blockinfo中的pvalue列
- tandem: 是否过滤由串联基因所形成的共线性区,即点阵图中对角线部分
- tandem_length: 如果过滤,那么评估tandem的标准就是两个区块的物理距离
- block_length: 一个共线区的最小基因对的数量,对应blockinfo中的length列
最后运行wgdi,输出图片。
wgdi -bk ath.conf输出图片中的Ks的取值范围由参数area控制。图中的每个点都是各个共线性区内的基因对的Ks值。不难发现每个共线区内的Ks的颜色都差不多,意味着Ks值波动不大,基于这个现象,我们在判定Ks峰值的时候,采用共线性区的中位数就比其他方式要准确的多。在图中,我们大致能观察到2种颜色,绿色和蓝色,对应着两次比较近的加倍事件。
Ks频率分布图
除了用点阵图展示Ks外,我们还可以绘制Ks频率的分布情况。假如一个物种在历史上发生过多倍化,那么从那个时间点增加的基因经过相同时间的演化后,基因对之间的Ks值应该相差不多,即,归属于某个Ks区间的频率会明显高于其他区间。
首先,创建配置文件(-kp, ksPeak)
wgdi -kp ? >> ath.conf然后,修改配置文件
[kspeaks]
blockinfo = ath_block_information.csv
pvalue = 0.05
tandem = true
block_length = 5
ks_area = 0,10
multiple = 1
homo = -1,1
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = ath.ks_median.distri.pdf
savefile = ath.ks_median.distri.csv这一步除了输出Ks峰图(savefig)外,还会输出一个根据输入文件( blockinfo )进行过滤后的输出文件(savefile)。过滤标准除了之前Ks点阵图提及的 tandem, pvalue, block_length 外,还多了三个参数, ks_area, multiple, homo.
pvalue: 共线性区的显著性, 对应blockinfo中的pvalue列,pvalue设置为0.05时会保留看着很不错的共线性片段,但是会导致古老片段的减少。
ks_area对应blockinfo中的ks列,该列记录了共线区所有基因对的ks值。ks_area=0,10 表示只保留ks值在0到10之间的基因对。
multiple和blockinfo中的homo1,homo2,homo3,home4,homo5...列有关。一般默认为1, 表示选择homo1列用于后续的过滤。如果改成multiple=2, 表示选择homo2
homo用于根据共线性中基因对的总体得分(取值范围为-1到1,值越高表明最佳匹配的基因对越多)对共线性区域进行过滤。当multiple=1, homo=-1,1时,表示根据homo1进行过滤,只保留取值范围在-1到1之间的共线性区。
最后运行程序
wgdi -kp ath.conf
从图中,我们能够更加直观的观察到2个peak,基本确定2个多倍化事件。
既然得到了一个Ks的peak图,我们可以和另一款工具wgd的拟南芥分析结果进行对比。wgd的Ks值计算流程为,先进行所有蛋白之间的相互比对,根据基因之间的同源性进行聚类,然后构建系统发育树确定同源基因,最后调用PAML的codeml计算Ks,对应下图A的A.thaliana。如果存在参考基因组,那么根据共线性锚点(对应下图D)对Ks分布进行更新, 对应下图A的A.thaliana anchors。
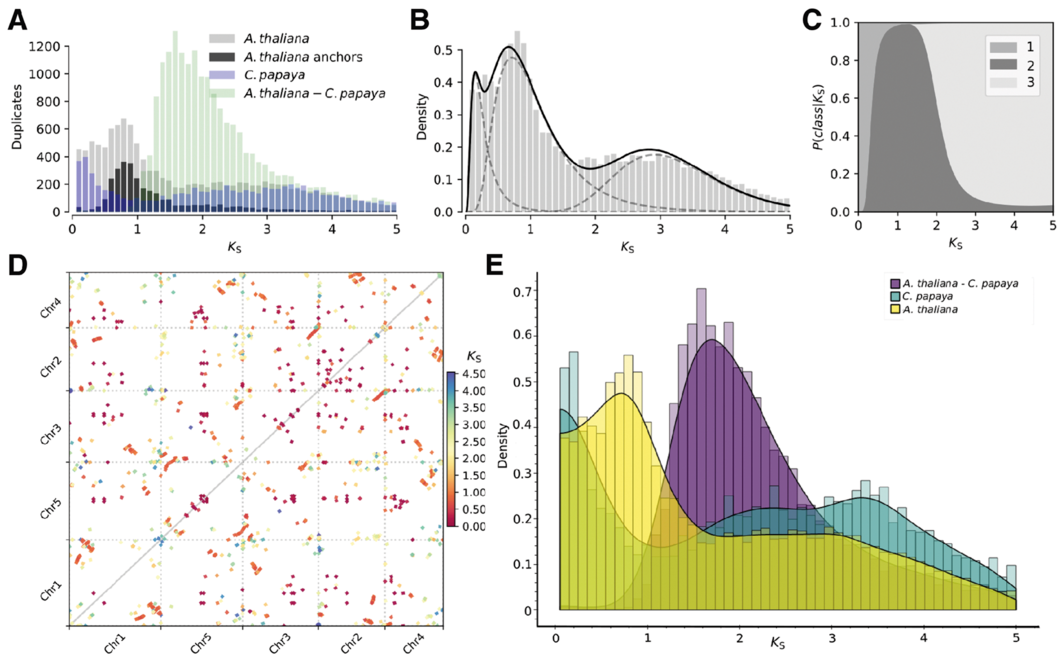
同样是拟南芥的分析,wgd的点图(上图D)信息比较杂乱,存在较多的噪声点,而WGDI的Ks点阵图能有效的反应出不同共线性区域的Ks特点。wgd的Ks分布图中的只能看到一个比较明显的峰,而WGDI的分析结果能明显的观察到两个。wgd在Ks上存在的问题很大一部分原因是它们是直接采用旁系同源基因计算ks,容易受到串联重复基因积累的影响。而wgd则是基于共线性区计算Ks,结果更加可靠,尤其是后续还可以通过不断的调整参数,来确保Ks的峰值正确判断,这也是为什么在绘制Ks频率分布图的同时还要生成一个过滤后的文件。
题外话: wgd是我在2019年学习WGD相关分析找到的一个流程工具,那个时候虽然也看了文章里面关于软件的细节介绍,但是由于对比较基因组学这一领域并不熟悉,所以也不好评判结果的可靠性。最近在学习wgdi时,一直和开发者反复讨论软件的一些参数细节,这才知道这里面的很多细节。这也警醒我,不能追求软件数量上的多,只求用软件跑出自己想要的结果发表文章,失去了科研的严谨性。
我们会在下一节介绍如何利用Ks点阵图和Ks频率分布图更可靠的拟合Ks峰值。

















 7941
7941

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









