







 PAGA(Partition-based Graph Abstraction)是一种新的图抽象方法,旨在通过生成保留数据全局拓扑的简化图来统一单细胞数据的聚类分析和轨迹推断。这种方法解决了现有算法在处理细胞异质性和连续过程时的挑战,如发育和疾病进展。PAGA通过分析细胞群的连接性,生成一个可解释的图映射,允许在不同分辨率下分析数据,提高探索性数据分析的效率。在多个造血数据集和生物系统中,PAGA表现出对复杂细胞谱系关系的准确预测和稳健的拓扑重构能力。
PAGA(Partition-based Graph Abstraction)是一种新的图抽象方法,旨在通过生成保留数据全局拓扑的简化图来统一单细胞数据的聚类分析和轨迹推断。这种方法解决了现有算法在处理细胞异质性和连续过程时的挑战,如发育和疾病进展。PAGA通过分析细胞群的连接性,生成一个可解释的图映射,允许在不同分辨率下分析数据,提高探索性数据分析的效率。在多个造血数据集和生物系统中,PAGA表现出对复杂细胞谱系关系的准确预测和稳健的拓扑重构能力。
PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells
PAGA:通过生成单细胞的拓扑结构并保存其映射来统一聚类和轨迹推断的图抽象
F. Alexander Wolf1 , Fiona K. Hamey2, Mireya Plass3, Jordi Solana3, Joakim S. Dahlin2,4, Berthold Göttgens2, Nikolaus Rajewsky3, Lukas Simon1 and Fabian J. Theis1,5*

Abstract:
ScRNA-seq可以量化离散细胞类型和连续细胞转变过程中的生物异质性。基于分区的图抽象(PAGA)在估计流形分区连通性的基础上,产生了流形数据的可解释类图映射。 PAGA映射保留了数据的全局拓扑,允许以不同的分辨率分析数据,从而提高了一般探索性数据分析工作流的计算效率。我们通过推断具有一致拓扑的四个造血数据集、成年扁平虫和斑马鱼胚胎的复杂结构细胞映射和在100万个神经元benchmarkPAGA的计算表现来说明该方法。 (https://github.com/theislab/paga)
Background:
ScRNA-seq为数千个单个细胞的全面分子分析提供了无与伦比的机会,预期将对广泛的生物医学研究领域产生重大影响。由此产生的数据集经常用术语转录景观(landscape)讨论。然而,对这些景观的细胞异质性和模式的算法分析,仍然面临着根本性的挑战,例如,如何解释细胞到细胞的变化(cell-to-cell variation),目前的计算方法通常以如下两种方式之一来解决这一点 [1]。聚类假设数据由生物学上不同的分组组成,例如不同的细胞类型或细胞状态,用不同的变量(聚类索引)给这些分组打标签。 相比之下,推断细胞的伪时序或轨迹[2-4]是假设数据位于连接的流形上,并用连续变量(沿流形的距离)来给细胞打标签。虽然前者是大多数单细胞数据分析的基础,然而后者可以更好地解释连续表型和过程,例如发育,剂量反应和疾病进展。 本文则将这两种观点统一。
在单细胞实验中一个很重要的例子是分析细胞异质性,分析细胞异质性会涉及到源自复杂细胞分化过程的数据。但是,使用伪时序[2,5–9]分析此类数据,通常面临着生物过程是不完全采样的问题。因此,实验数据不符合连通流形,这样将数据建模成连续树形结构(现有算法的基础)是没什么意义的。这个问题甚至存在于基于聚类的,推断树形结构的算法过程中[10-12],它通常做出聚类符合一个连通的树形拓扑的无效假设。而且,它们依赖于基于聚类间距离的特征空间,例如聚类均值的欧式距离。然而,这种距离度量只在局部尺度上量化细胞的生物相似性,并且在用于更大规模的事物(如聚类)时充满了问题。通过采样[11,12]来解决拟合树形到聚类之间距离的高度不稳健[10]的努力效果有限。
基于分区的图抽象(PAGA)解决这些基本问题是通过生成细胞的类图映射,这可以保存多种分辨率下的数据中的连通和不连通结构。PAGA的数据分析公式可以跨不同的数据集,稳健地重建分支上基因表达的变化,并且第一次重建整个成年动物[13]的谱系关系。此外,PAGA初始化的流形学习算法收敛得更快,产生的嵌入更忠于高维数据的全局拓扑,并引入了一种基于熵的度量来量化这种忠实性。最后,展示了PAGA如何抽象转变图,比如,来自RNA速率,并与以往的轨迹推断算法进行了比较。因此,PAGA提供了一种图抽象方法[14],适用于推导有噪声的KNN图(通常用于表示scRNA-seq数据产生的流形)的可解释抽象。
Results:
PAGA映射离散不连通和连续连通的细胞到细胞的变化
现有的流形学习技术和单细胞数据分析技术都将数据表示为单细胞G=(V,E)的邻域图,其中V中的每个节点对应于一个细胞,E中的每个边表示邻居关系 (Fig. 1) [3, 15–17]。然而,G的复杂性和噪声相关的假边使得很难追溯从祖细胞到不同命运(fates)的一个假定的生物过程,也很难决定细胞组之间,实际上是连接的还是断开的。此外,追溯单细胞的孤立路径来陈述生物过程,其统计能力太少从而无法达到可接受的置信水平。 通过对单细胞路径的分布进行平均来获得power受到了拟合这些路径的分布的实际模型的困难的阻碍。
我们通过开发一个关于细胞群连接性的统计模型来解决这些问题,细胞群通常通过图分区[17-19]或通过聚类或实验注释来确定。这生成一个更简单的PAGA图G∗(Fig. 1),其节点对应于细胞群,其边权量化群之间的连通性。与模块化[20]类似,统计模型认为,如果细胞群之间的边数超过随机分配下期望边数的一部分,则群之间就是连接的。连接强度可以解释为对实际连接存在的可信度,并允许丢弃与噪声相关的,假的连接(Additional file 1:Note 1)。
当G表示数据在单细胞分辨率下的连通性结构时,PAGA图G∗表示数据在选定粗分辨率的分区的连通性结构,并允许识别数据的连通和断开区域。沿着G ∗中的节点的路径意味着通过G中相应的细胞群的单细胞路径的集合。通过平均这样的单细胞路径的集合,有可能以对假边稳健的方式追溯从祖先到命运的假定生物过程,提供统计能力,并与细胞生物轨迹的基本假设一致(Additional file 1: Note 2)。注意,通过改变分区的分辨率,PAGA可以产生多个分辨率的图,从而能够对数据进行分层探索 (Fig.1Additional file 1:Note 1.3)。
为了追踪单细胞分辨率下的基因动力学,我们将现有的基于随机游走的距离测度(Additional file 1: Note 2, Reference [7])扩展到考虑了不连通图的现实际情况。通过追溯抽象图G ∗中的高置信度路径和根据该路径中每组中的细胞与祖细胞的距离d排序,我们以单细胞分辨率追踪基因变化 (Fig. 1)。因此,PAGA通过提供一个坐标系(G∗,d)来涵盖聚类和伪时序的两个方面,该坐标系允许我们在保持其拓扑的同时探索数据的变化 (Additional file 1: Note 1.6)。因此,PAGA可以被看作是一种易于解释和稳健的拓扑数据分析方法 [9, 21] (Additional file 1: Note 3)。
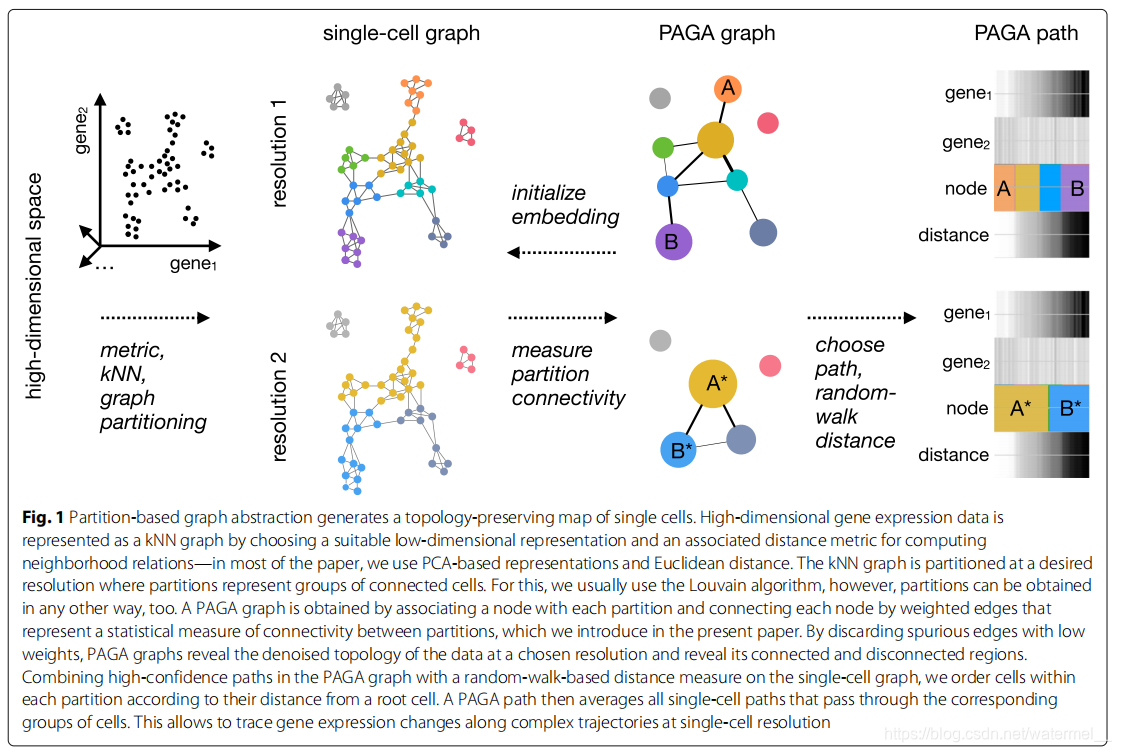
Fig1: 基于分区的图抽象生成保留拓扑的单细胞映射。高维基因表达数据通过选择合适的低维表示形式和相关的距离度量这是为了计算邻域关系(在大部分论文中)表示为KNN图,我们使用基于PCA的表示和欧几里德距离。将KNN图以期望的分辨率分区,其中分区表示连接的细胞的群(partitions represent groups of connected cells.)。 为此,我们通常使用Louvain算法,然而,分区也可以任何其他方式获得。通过将一个节点(G中的节点)与每个分区(对KNN图执行louvain得到的分区)相关联,并通过表示分区之间连通性的统计度量的加权边连接每个节点,得到一个PAGA图G,本文介绍了这一点。通过丢弃低权重的假边,PAGA图揭示了数据在选定分辨率下的去噪拓扑,并揭示了其连接和断开区域。将PAGA图中的高置信度路径与单细胞图上基于随机游走的距离度量相结合,我们排序每个分区内的细胞根据它们到根细胞的距离。然后,一个PAGA路径平均所有通过相应细胞组的单细胞路径。 这允许在单细胞分辨率下沿着复杂的轨迹跟踪基因表达的变化。
PAGA初始化的流形学习产生保留拓扑的单细胞嵌入
计算上几乎无成本的PAGA粗分辨率嵌入可以用来初始化已有的流形学习和绘图算法,如UMAP[22]和ForceAtlas2(FA)[23]。本文采用这种策略生成单细胞嵌入。与以往算法的结果相比,PAGA初始化的单细胞嵌入忠于全局拓扑,这大大提高了它们的可解释性。为了量化这种说法,我们从嵌入算法的分类角度出发,开发了成本函数KLgeo(Box 1 and Additional file 1: Note 4),该函数分别通过在高维空间和嵌入空间中合并沿数据流形表示的测地距离来得到对全局拓扑的忠实度。与此之外,PAGA初始化的流形学
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 3726
3726

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









