










今天给大家分享一篇今年4月份发表在Nature methods期刊上的单细胞蛋白组学数据处理框架—scPROTEIN。简单介绍以后会有相应的实现方法。
scPROTEIN 可以在统一框架中估计肽定量的不确定性、对蛋白质数据进行去噪、消除批次效应并编码单细胞蛋白质组特异性嵌入。我们证明 scPROTEIN 可有效用于细胞聚类、批次校正、细胞类型注释、临床分析和空间分辨蛋白质组学数据探索。
该算法框架解决的问题:
1、批次效应
批次效应阻碍了单细胞蛋白质组学数据的分析。这些批次效应可以是内部的,也可以是外部的。内部批次效应包括液相色谱批次效应和串联质谱标签批次效应。当整合来自多个数据采集的数据集时会出现外部批次效应,这些数据集使用了不同的样品制备方法和标记策略。其次,由于样品制备过程中的样品损失,并不是所有的组成肽内容都被输送到质谱仪,这对单细胞蛋白质组学来说是一个至关重要的问题。
2、数据缺失和噪声
肽注入质谱仪后,在数据依赖型采集模式中,肽的信号强度会受到其电离效率和母离子峰选择的影响,从而可能导致单细胞蛋白质组数据中出现噪声或缺失。
3、肽不确定性
对于自下而上(从肽段鉴定蛋白)的单细胞蛋白质组学,核心定量是在肽水平上。然而,现有的单细胞蛋白质组学数据处理流程并没有充分考虑这种层次内容。在实际应用中,现有的质谱采集技术在准确定量肽水平方面存在缺陷。因此,如果不考虑肽测量固有的不确定性而得出蛋白质含量,将会产生不准确的结果。为了提高构建蛋白质丰度数据的精度,建议采用为肽分配不确定性权重的方法来提高蛋白质丰度数据的准确性。这种补偿措施解决了与定量过程相关的固有不准确性。
该算法框架原理:
通过提供多功能细胞嵌入,在统一框架中解决肽定量的不确定性、数据缺失、批次效应和高噪声问题。首先,对于提供原始肽信号强度的数据集,我们提出了一个多任务异方差回归模型来估计肽定量的不确定性,并以不确定性引导的方式将肽含量聚合到蛋白质水平(图a)。然后,我们构建了一个图结构来表征单细胞蛋白质组学数据,其中考虑共表达模式的消息传递过程有助于缓解数据缺失问题。开发了一种具有设计交替拓扑属性去噪模块的图对比学习模型,该模型可以对蛋白质组数据进行去噪并实现准确的表示。此外,对比学习的判别性和去噪模块可以在不知道数据集的先验知识的情况下一起隐式地缓解批次效应(图b)。最后,学习到的多功能细胞嵌入可以应用于各种下游任务(即细胞聚类、批次校正和细胞类型注释)(图c)。
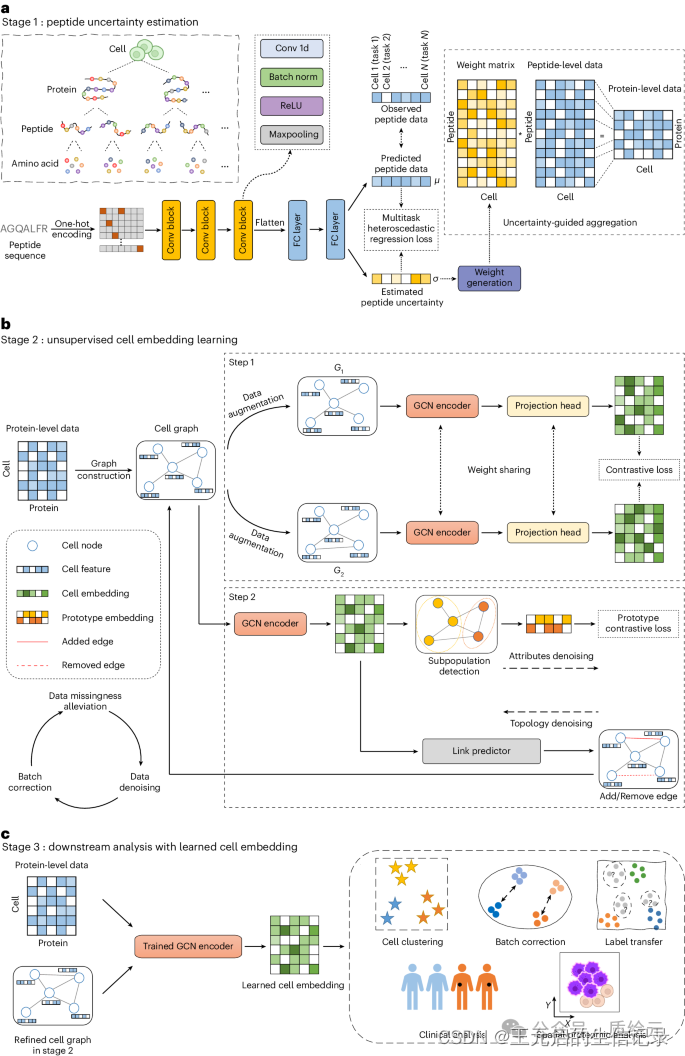
算法实现:
1、环境配置
#####python 环境
python>=3.8
#####直接pip安装,暂时没有报错,一次性安装成功
pip install scprotein
#####如果有报错,网络不好等情况,请下载.whl文件自行安装
pip install docs/scprotein-0.1.1-py3-none-any.whl
#####以下命令确认安装成功与否
python3 -c "import scprotein"
2、 命令行分析
#####第一阶段数据分析
python3 train_stage1.py
#####第二阶段数据分析
python3 train_stage2.py --stage1 True
#####第三阶段数据可视化
python3 visualization.py
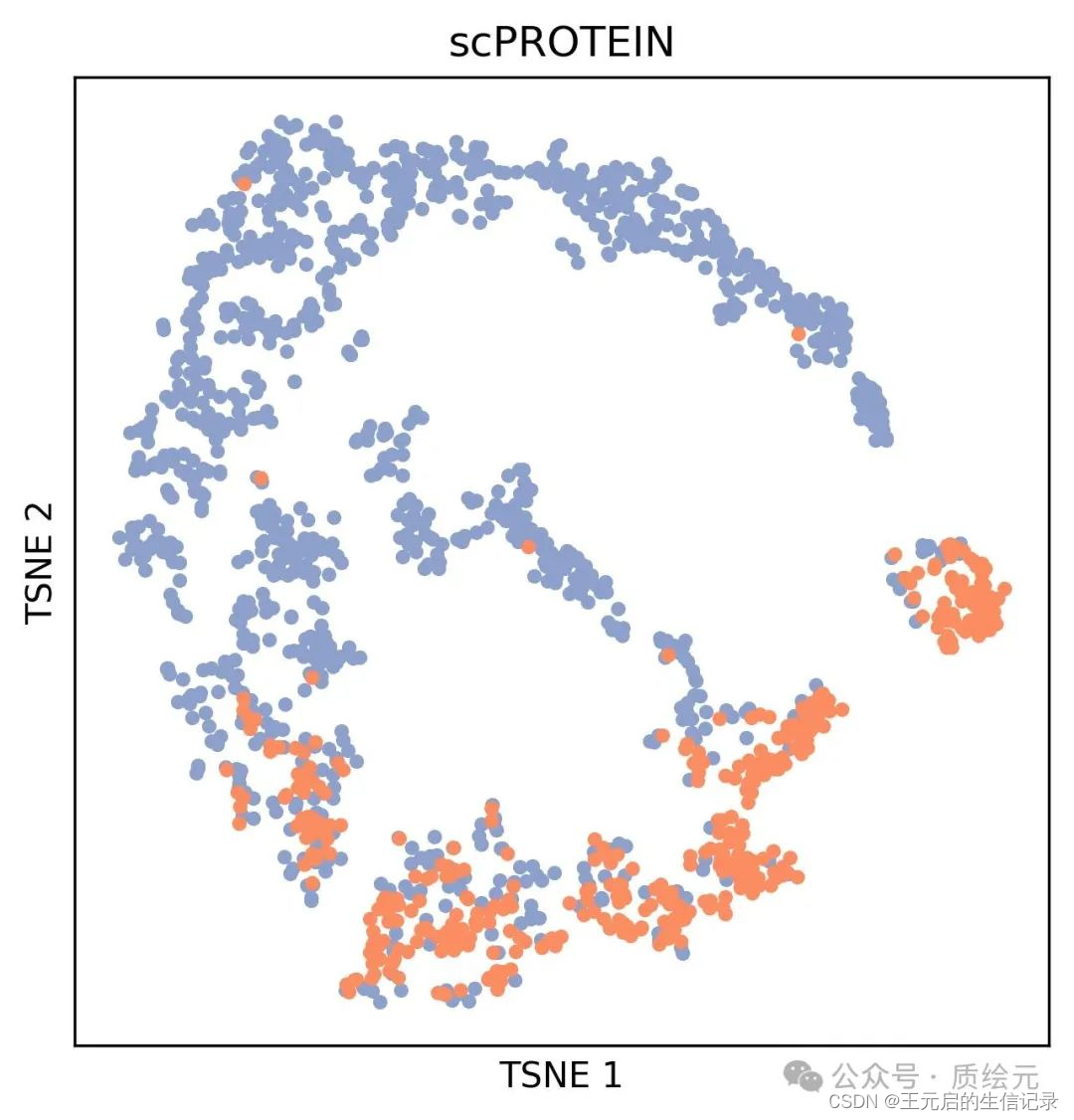
3、结果展示
经过第2步后,生成t-sne降维结果。
4、使用下载好的训练模型进行评估
#####loading checkpoints
python3 train_stage1.py --use_trained_scPROTEIN True
python3 train_stage2.py --stage1 True --use_trained_scPROTEIN True
已经测试过,8GB运行内存电脑可以进行分析,对结果还可以进行后续的分析。
该算法框架比较小巧好使用,欢迎大家测试分析,如有疑问,请联系管理员:kriswcyYQ进行解决。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









