






大家好,在转录组测序分析中,有三个经典的数值,即count,FPKM以及TPM值。在TCGA数据库中,其提供了count和FPKM两种结果形式。而平时的分析过程中,FPKM和TPM往往是我们比较常用的数据标准化方法。
首先,我们来简单看一下三者的基本概念。
count:原始测序得到的count数就是比对到某个基因i上的总数目;不知道大家是否了解测序的简单过程?在测序分析过程中,我们首先是将测得的短reads比对到参考基因组上,然后通过软件来计算该片段上比对到reads的数量,也就是说呢,count是一个整数值。
FPKM:我们把比对到的某个基因i的Fragment数目,除以基因的长度,其比值再除以所有基因的总长度。注意,严格来讲,这里的基因长度是指基因i外显子的总长度。
TPM:与FPKM不同的地方在于,其基因的比值是再除以(基因的总数目/基因的总长度)。因此,其得到的结果是一个相对的比值。
比较三者的定义,我们可以发现,FPKM和TPM两种标准化方法的计算公式,其分子是完全相同的,唯一的区别在于对于分母处的处理方式。如果已知FPKM的话,那么TPM的值就是可以通过FPKM除以FPKM值的总和,再乘以10的6次方而得到。相对来讲,就标准化程度而言,FPKM值是不如TPM值的,因此在后续的分析过程中我们一般是推荐使用TPM值的。
下面,我们将介绍一下如何使用R语言来进行count,FPKM和TPM三者之间的转换。
1.基因长度的计算
首先,我们来看一下基因Length的计算方法。相信大家必然听说过可变剪切的概念,也正是因为可变剪切的存在,同一个基因会产生不同的转录本。在这里呢,又会产生两种不同的分析思路:
思路1:计算基因在染色体的起始和结束之差;
思路2:计算每个基因的最长转录本或所有外显子之和;
那么,又有一个问题产生了,如何去获取基因在染色体上的位置信息呢?对基因组测序或分析有些了解的小伙伴应该对这几个文件类型有所接触,想fasta文件是保存基因DNA或RNA的测序信息,gtf、gff以及gf3文件则都是保存基因注释的文件。我们来一起看一下基因注释文件里面所包含的内容:
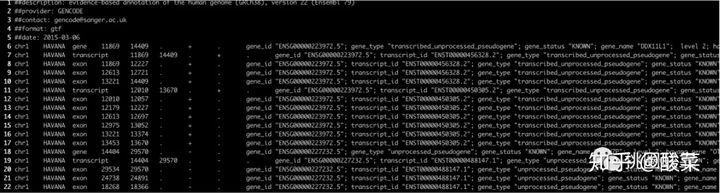
在该文件中,我们可以看到,这是一份人类参考基因组注释文件,GRCH38,版本是22,存在于GeneCode网站上,当然现在已经有相应的更新版本;里面包含了基因的染色体位置,起始和终止位置;
根据思路1,两位置相减即为基因长度。同时,+代表其为正向,-则代表为反向,其长度需要后面位置减去前面位置。往后是基因注释的id号,比如Ensemble号,symbol号等具体信息。
接着,我们来演示一下如何用R来进行计算,这里使用的R包是biomaRt包。

加载完这个R包后,通过listMarts()函数查看可以连接到的 BioMart 数据库;

首先来查看一下基因组的参数,在此我们使用的是Ensemble id号;通过useMart()函数设置要连接的 BioMart 数据集。

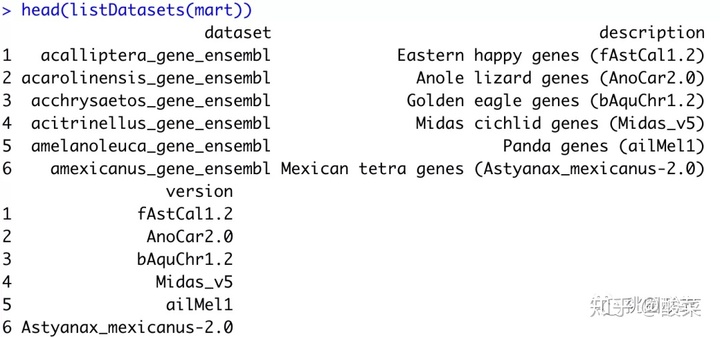
随后,这里以人类基因组为例,获取基因组信息;注意不同的物种之间是不一样的;

getBM()函数是检索数据用到的主要函数,首先需要对它的4个参数 (filters, attributes, values, mart) 及参数选项进行查看和选择。然后,我们从输入数据中提取基因名。

接着,我们对每个基因计算有效长度eff_length;

取染色体上位置相减的绝对值作为每个基因的长度,最终得到了第1种思路的基因长度结果。
而对于第2种思路来讲,其存在不同的转录本,同一个基因就可以存在多个转录本,分别相减取其中最长的一个,或者将这些外显子分别计算,再进行相加从而得到结果。

接着,导入GTF 或者GFF3文件,ensembl或者gencode网站GTF注释皆可;在此,我已经将文件进行了下载;

读取gtf文件,并通过exonBy()函数提取基因的外显子信息;
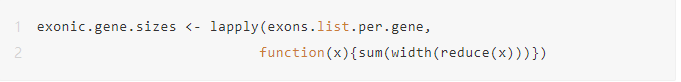
同时,我们进一步可以通过reduce()函数来避免重复计算重叠区;
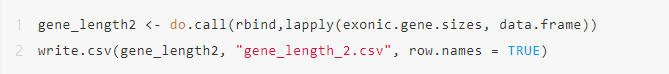
并且,对生成的基因长度结果赋予geneID,即ensemble编号;这样一份完整的基因长度文件就准备完成了。
2.Count值转换成FPKM值
随后,我们来对示例数据中的count值进行转换。
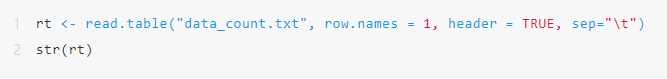
在count文件中,一共包含了6例样本,56830个不同的基因表达。

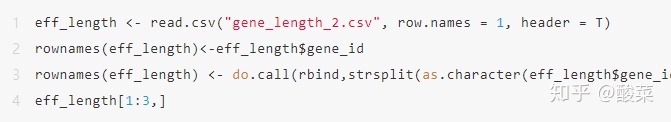
接着,将之前准备好的基因长度文件进行读取;这里,我们选取第二种方法计算得到的基因长度文件。


在正式的转换之前,我们需要将表达文件和基因长度文件做一个预处理。取共同基因,使得两者的基因名字和排序一致。最终一共得到了56830个共同基因。

根据转换的计算公式,设置一个新的函数,并结合apply()函数,对其直接进行转换,最终得到了每个基因对应的FPKM值。
3.FPKM值转TPM值
接下来,同样的,设置了FPKM值转换成TPM值的函数。

这样,count值,FPKM值和TPM值之间的转换就完成了。在单基因分析模式中,一般还是推荐使用TPM值进行后续的分析计算。
作者:阿琛
本文首发于“ 解螺旋”微信公众号
转载请注明:解螺旋·临床医生科研成长平台















 7083
7083











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









