






FPKM: Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments)
FPKM:Fragments per Kilobase Million,FPKM意义与RPKM极为相近。二者区别仅在于,Fragment 与 Read。RPKM的诞生是针对早期的SE测序,FPKM则是在PE测序上对RPKM的校正。只要明确Reads 和 Fragments的区别,RPKM和FPKM的概念便易于区分。Reads即是指下机后fastq数据中的每一条Reads,Fragments则是指每一段用于测序的核酸片段,在SE中,一个Fragments只测一条Reads,所以,Reads数与Fragments数目相等;在PE中,一个Fragments测两端,会得到2条Reads,但由于后期质量或比对的过滤,有可能一个Fragments的2条Reads最后只有一条进入最后的表达量分析。总之,对某一对Reads而言,这2条Reads只能算一个Fragments,所以,Fragment的最终数目是Reads的1到2倍之间。
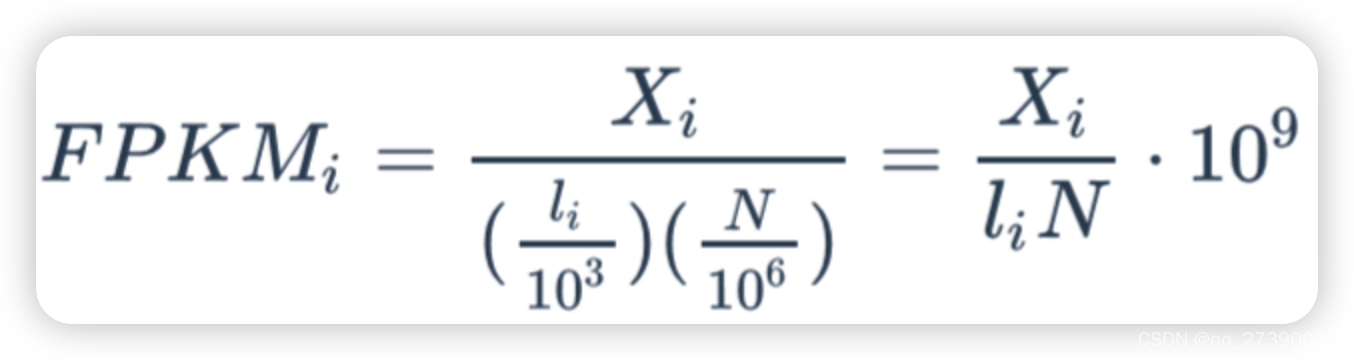
![]()
TPM:Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts)

1. counts 转 FPKM
计算FPKM的三要素:原始counts矩阵,样本总reads数,基因长度。
# expr: counts 表达矩阵, 行:转录本,列:样本
# transcript_len$length 转录本长度
expr1 = expr/transcript_len$length
fpkm = t(t(expr1)/colSums(expr)) * 10^92. counts 转 TPM
# expr: counts 表达矩阵
# transcript_len$length 转录本长度
expr1 = expr/transcript_len$length
fpkm = t(t(expr1)/colSums(expr)) * 10^9
tpm <- t(t(fpkm)/colSums(fpkm))*10^6
## 函数法
Counts2TPM <- function(counts, effLen){
rate <- log(counts) - log(effLen)
denom <- log(sum(exp(rate)))
exp(rate - denom + log(1e6))}
tpm <- Counts2TPM(expr,transcript_len$length)3. FPKM转TPM
tpm = t(t(fpkm)/colSums(fpkm))*10^6
## 函数法
# FPKM数据转为TPM数据
FPKM2TPM <- function(fpkm){
exp(log(fpkm) - log(sum(fpkm)) + log(1e6))}
tpm <- apply(fpkm,2,FPKM2TPM)limma,edgeR, DESeq2进行差异表达分析,要用原始counts矩阵。
FPKM可以用limma去运行,不过数值比较大的话要先进行log2转化。

















 9226
9226

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









