






在生命科学的广阔领域中,蛋白质与小分子配体之间的相互作用扮演着至关重要的角色。这些相互作用不仅影响着生物体内的各种生命活动,如信号传导、代谢调控和药物作用等,同时也是药物设计和开发的核心内容。因此,深入理解并模拟这些相互作用过程,对于推动生命科学研究和药物研发具有重要意义。
本教程旨在为读者提供一套完整的蛋白质与小分子配体相互作用模拟的流程和方法。通过本教程的学习,您将能够掌握从蛋白质与小分子配体的结构准备、相互作用模拟到结果分析的全流程,从而能够自主进行相关的模拟研究。
在本教程中,我们将首先介绍蛋白质与小分子配体相互作用的基本原理和模拟的基本概念,为读者奠定理论基础。随后,我们将详细阐述模拟的具体步骤,包括结构准备(如蛋白质结构预测、小分子结构优化等)、相互作用模拟(如分子对接、分子动力学模拟等)以及结果分析(如相互作用能计算、轨迹分析等)。在每个步骤中,我们都会结合具体的案例和实例,详细解释操作步骤和注意事项,帮助读者更好地理解和掌握。
具体流程:
一、预处理复合物
1. 蛋白质及配体结构获取
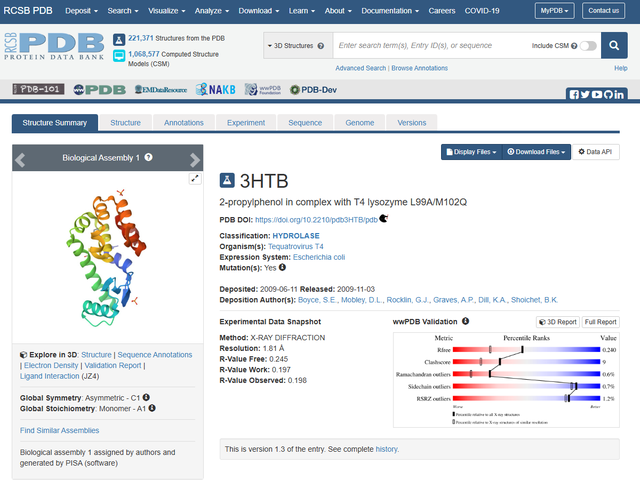
在本教程中,我们将使用T4溶菌酶L99A/M102Q(PDB ID:3HTB)为例,从PDB蛋白数据库 (RCSB PDB)下载其晶体结构,去掉晶体水,PO4和 BME。
- 蛋白及配体力场获取
只有在力场的.rtp文件中存在构建块的条目时,拓扑才能自动组装。而JZ4配体在 GROMACS 提供的任何力场中都不是一个可识别的实体,因此我们将分两步准备系统拓扑:1)用pdb2gmx准备蛋白质拓扑;2)使用外部工具准备配体拓扑。
2.1 使用pdb2gmx准备蛋白质拓扑
本教程使用的力场为amber14sb.ff,选择默认的水模型TIP3P,然后为封端选择“NH3+”和“COO-”,获得力场文件及完整坐标文件。
2.2 使用外部工具获得配体拓扑
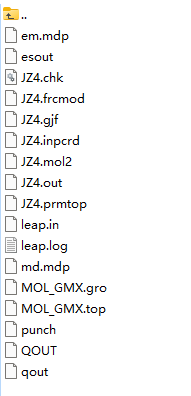
本教程使用的力场为amber14sb.ff,因此使用GAFF工具生成JZ4配体的top文件。1)使用Avogadro软件为配体添加氢原子,同时输出JZ4.com文件,修改Gaussian设置,获得Gaussian输入文件JZ4.gjf,进行 Gaussian 优化。2)利用 AmberTools 计算电荷。3)利用 parmchk 检查成键相缺失。4)利用LEaP生成 Amber 格式力场,文件内容见下图。5)利用acpype将Amber格式转换为Gormacs格式的GAFF力场文件及坐标文件。

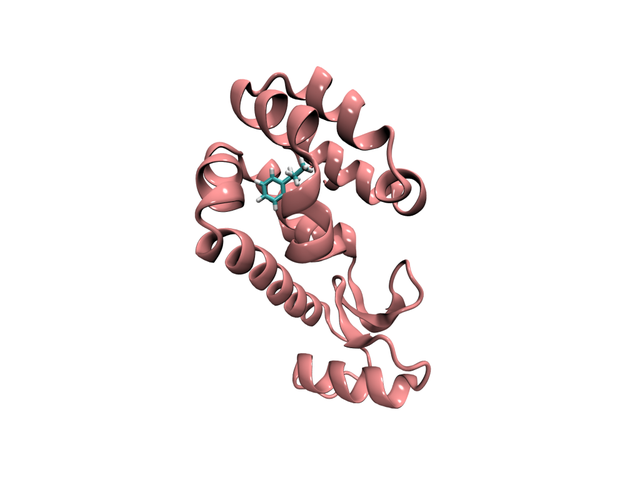
- 组合蛋白质和配体,生成蛋白质-配体复合物
二、定义盒子,添加溶剂及离子
三、能量最小化
四、限制复合物及体系平衡
1. 限制复合物:通过genrestr创建位置限制文件,定义位置限制。
2. NVT平衡
3. NPT平衡
五、成品模拟
六、分析
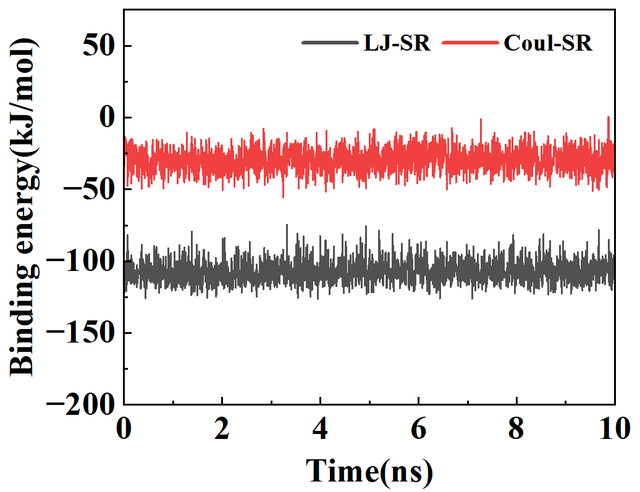
1. 执行energy模块计算蛋白质-配体相互作用
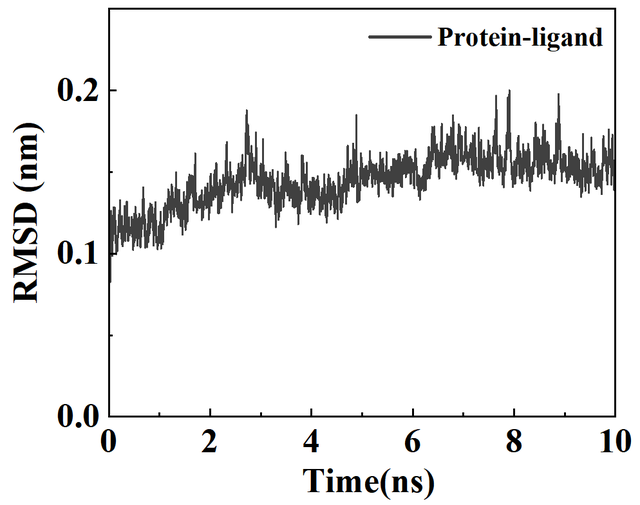
2. 执行rms模块,计算RMSD
公众号“320科技工作室”















 4873
4873

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









