






准备输入文件
step 1 利用autodock对接蛋白质和小分子
step 2 利用pymol 输出 protein结构为pdb格式,输出ligand为mol2
step 3 处理蛋白质和小分子获得gro itp top文件用于Gromacs分子动力学模拟
蛋白质的拓扑文件获取
- gromacs 能够直接处理蛋白质获得gro itp top
gmx pdb2gmx -ignh -ff amber99sb-ildn -f protein.pdb -o protein.gro -p protein.top -water tip3p
小分子top文件获得
小分子的top文件获得与蛋白质top文件获得相比要难,gromacs不能直接处理小分子,需要用到其他的软件。(autodocktools, sobtop)
autodocktools能够对小分子进行加氢处理.
可以利用sobtop软件生成小分子的top gro
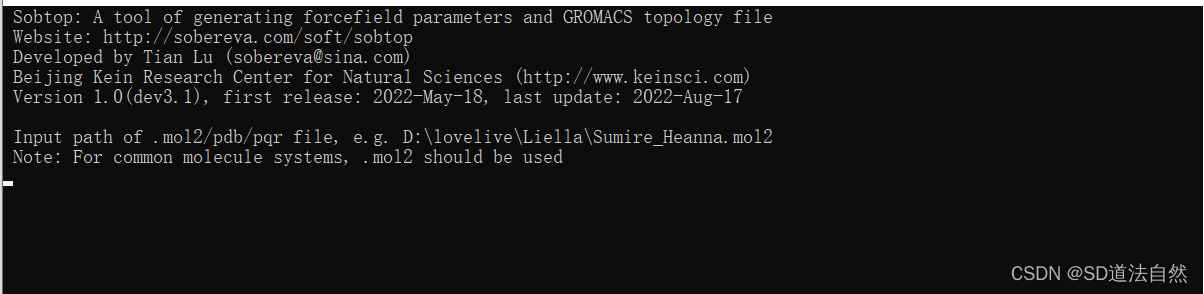
sobtop界面 首先输入ligand的文件路径(下载sobtop文件然后解压,点击sobtop.exe)(http://sobereva.com/soft/Sobtop/#download)
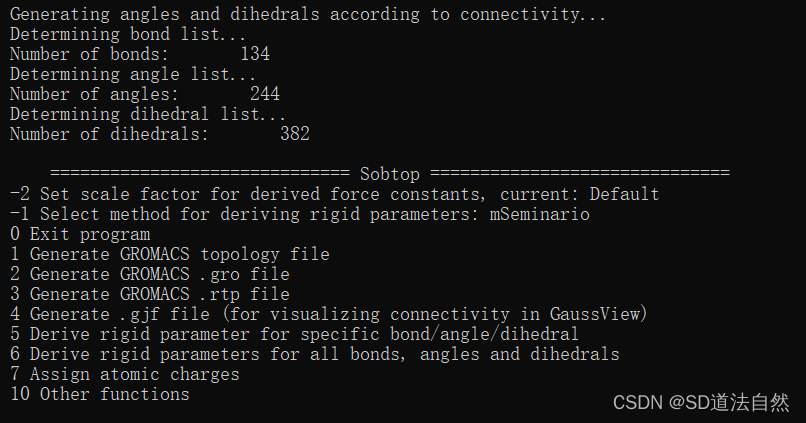
进入选择界面,首先生成小分子的top文件 选择 1
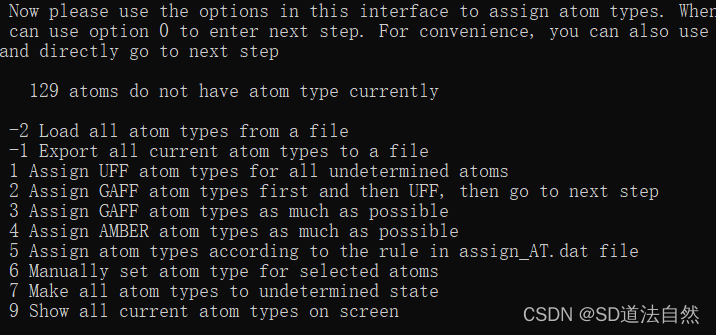
这里提示用什么样的力场来处理小分子,这里选择 2
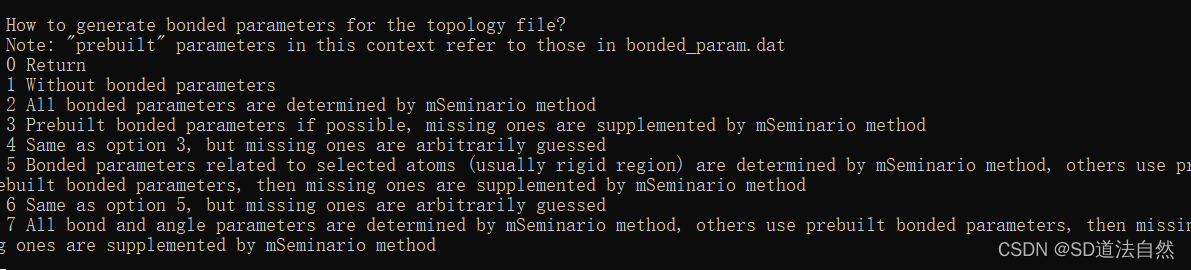
选择什么样的参数来处理小分子的键相互作用 选择4
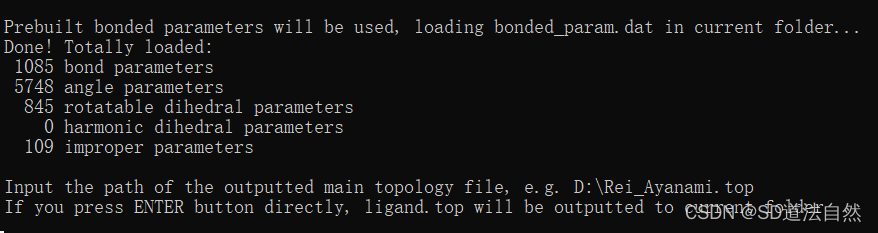
这里需要输入 输出的文件路径 .top /itp

选择2 生成gro 文件
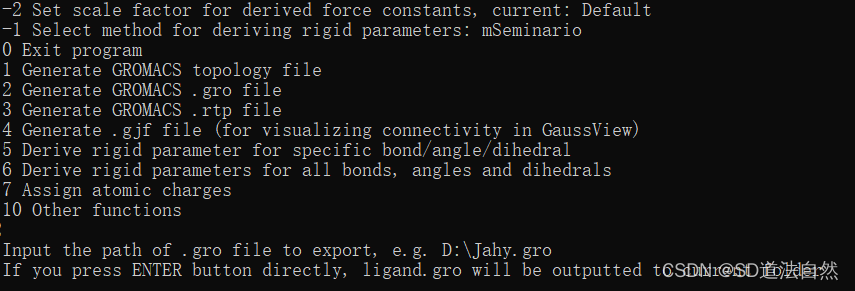
输入输出gro文件的路径
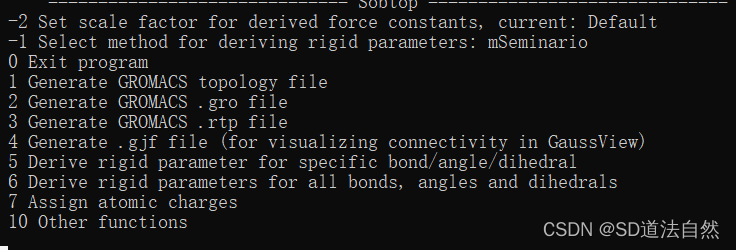
选择 0 推出即可
到这里我们就获得了protein和ligand的gro top itp文件,接下来合并protein和ligand的gro,top文件
step1 gro的合并
创建一个空的文件命令为complex.gro
将protein.gro的文件先复制进去,然后在protein.gro的倒数第二行后,将ligand.gro复制进去,
更改 第二行的原子数目,保存
step 2 top的合并
创建一个空的文件命令为complex.top
将protein.top的文件复制进去.
这里我们需要去修改力场的 ,因为小分子的原子类型是不包括在本身的力场中的,我们需要对力场的原子类型进行更改,找到生成protein的力场(力场文件在Gromacs安装包下share文件夹中)重新复制一份,将小分子itp文件中的 atomtype 下的原子类型复制到 力场文件中 ffnonbonded.itp 中的atomtype下,并删除小分子itp文件中的 atomtype。
然后将protein. top 中的
; Include forcefield parameters
#include “amber99sb-ildn.ff/forcefield.itp”
; Include water topology
#include “amber99sb-ildn.ff/tip3p.itp”
; Include topology for ions
#include “amber99sb-ildn.ff/ions.itp”
都修改成对应的力场文件
; Include forcefield parameters
#include “xx.ff/forcefield.itp”
; Include water topology
#include “xx.ff/tip3p.itp”
; Include topology for ions
#include “xx.ff/ions.itp”
并把小分子的top包括protein.top中
; Include ligand
#include “ligand.itp”
并把小分子的top中的
default 去掉
并把 complex.top 文件中的
[ molecules ]
; Compound #mols
Protein_chain_A 1
ligand 1 加上
然后进行下一步
#2.创建盒子
gmx editconf -f complex.gro -o complex.gro -bt dodecahedron -d 1
#3.蛋白质分子真空中的能量最小化
gmx grompp -f mdp/em-vac-pme.mdp -c complex.gro -p 5g2x.top -o em-vac.tpr -maxwarn 2 -r complex.gro
#使用mdrun命令运行能量最小化
gmx mdrun -v -deffnm em-vac
#4.向盒子中填充溶剂及离子并进行能量最小化
#添加溶剂离子
gmx solvate -cp em-vac.gro -cs spc216.gro -p 5g2x.top -o 5g2x-b4ion.gro
#生成 tpr文件
gmx grompp -f mdp/em-sol-pme.mdp -c 5g2x-b4ion.gro -p 5g2x.top -o ion.tpr -maxwarn 2 -r 5g2x-b4ion.gro
#添加离子模拟体内环境
#add ions neutral (中和电荷)
gmx genion -s ion.tpr -o ion.gro -neutral -conc 0.05 -p 5g2x.top
#再次生成tpr文件
gmx grompp -f mdp/em-sol-pme.mdp -c ion.gro -p 5g2x.top -o em-sol.tpr -maxwarn 2 -r ion.gro
#使用mdrun命令运行能量最小化
gmx mdrun -v -deffnm em-sol
#第六步: 位置限制性预平衡模拟
#预处理文件, 并运行NVT模拟
gmx grompp -f mdp/nvt-pr-md.mdp -r em-sol.gro -c em-sol.gro -p 5g2x.top -o nvt-pr.tpr -maxwarn 2
gmx mdrun -deffnm nvt-pr
#预处理文件, 并运行NPT模拟
gmx grompp -f mdp/npt-pr-md.mdp -c nvt-pr.gro -p 5g2x.top -o npt-pr.tpr -r nvt-pr.gro -maxwarn 2
gmx mdrun -deffnm npt-pr
#7.成品模拟
gmx grompp -f mdp/npt-nopr-md.mdp -c npt-pr.gro -p 5g2x.top -o npt-nopr.tpr
gmx mdrun -deffnm npt-nopr -v --updata gpu -nptmpi 1
#############运行完后分析###################################################
RMSD
gmx rms -s npt-nopr.tpr -f npt-nopr.trr -o fws-protein-rmsd.xvg # protein
gmx rms -s npt-nopr.tpr -f npt-nopr.trr -o fws-ligand-rmsd.xvg # ligand
RMSF
gmx rmsf -s npt-nopr.tpr -f npt-nopr.trr -b 500 -o fws-rmsf.xvg -ox fws-avg.pdb -res
gyrate
gmx gyrate -s npt-nopr.tpr -f npt-nopr.trr -o fws-gyrate.xvg

















 718
718

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









