






 本文详述了使用Materials Studio进行材料编辑,包括分子操作、超胞构建、表面切割、添加真空层以及石墨烯的制备方法。通过调整Display Style、运用Symmetry、Supercell和Build Vacuum Slab等功能,介绍了如何创建和修改材料结构,特别是TiO2-Rutile、石墨烯和MoS2等材料的处理。此外,还提及了能带计算的初步步骤。
本文详述了使用Materials Studio进行材料编辑,包括分子操作、超胞构建、表面切割、添加真空层以及石墨烯的制备方法。通过调整Display Style、运用Symmetry、Supercell和Build Vacuum Slab等功能,介绍了如何创建和修改材料结构,特别是TiO2-Rutile、石墨烯和MoS2等材料的处理。此外,还提及了能带计算的初步步骤。
此文根据山东大学魏巍老师的课堂讲课总结记录而成。
操作界面介绍
右键按住是旋转
多选,删除,复制,剪切,粘贴支持windows快捷键操作
选中后,同时按住shift+alt键可以快速对分子进行移动
绘制过程中,按住shift+左键单击变成选择
shift+右键长按,对选中单元进行旋转
File
新建导出另存为打印之类的操作
Edite
对图形中的剪切,粘贴,复制类操作
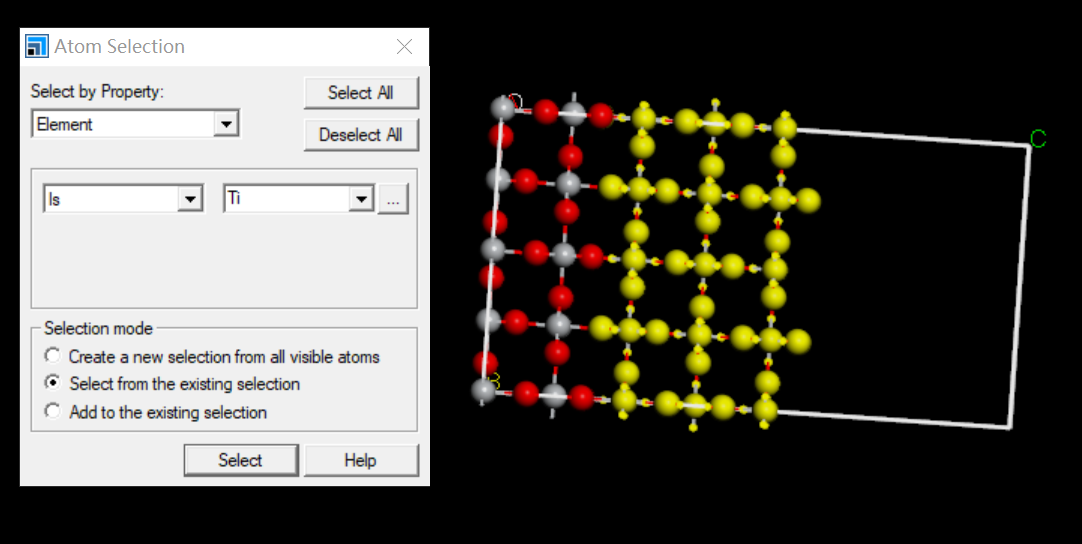
其中有一个Edit->Atom Selection,可以全图批量选中某种原子,或从选中的原子中批量选出某原子
从选中的原子中筛选出某元素的设置为

View
控制软件界面上显示的快捷功能,工具条,窗口,
你的MS和别人的显示界面不一样,可以尝试在这里开启不同的功能
例如,View->Explorers->Properties Explorer可以添加属性窗口
一些快捷工具View->Toolbars
 左键是其自身功能(选择,旋转,放大/移动)
左键是其自身功能(选择,旋转,放大/移动)


左单击是新建(单个原子,环,链),
左单击后,按ESC可实现添加原子
左键点住已有原子,可以拖动原子位置
单击已有原子/刚添加一个原子 后 左键长按可以添加一个新原子并和之前的原子成键

对图形的修改,测量距离角度,拟合,自动调整键的角度,补H,替换原子类型,改变键的类型,杂化类型
其他的一些工具条

一些窗口 VIew->Explorers
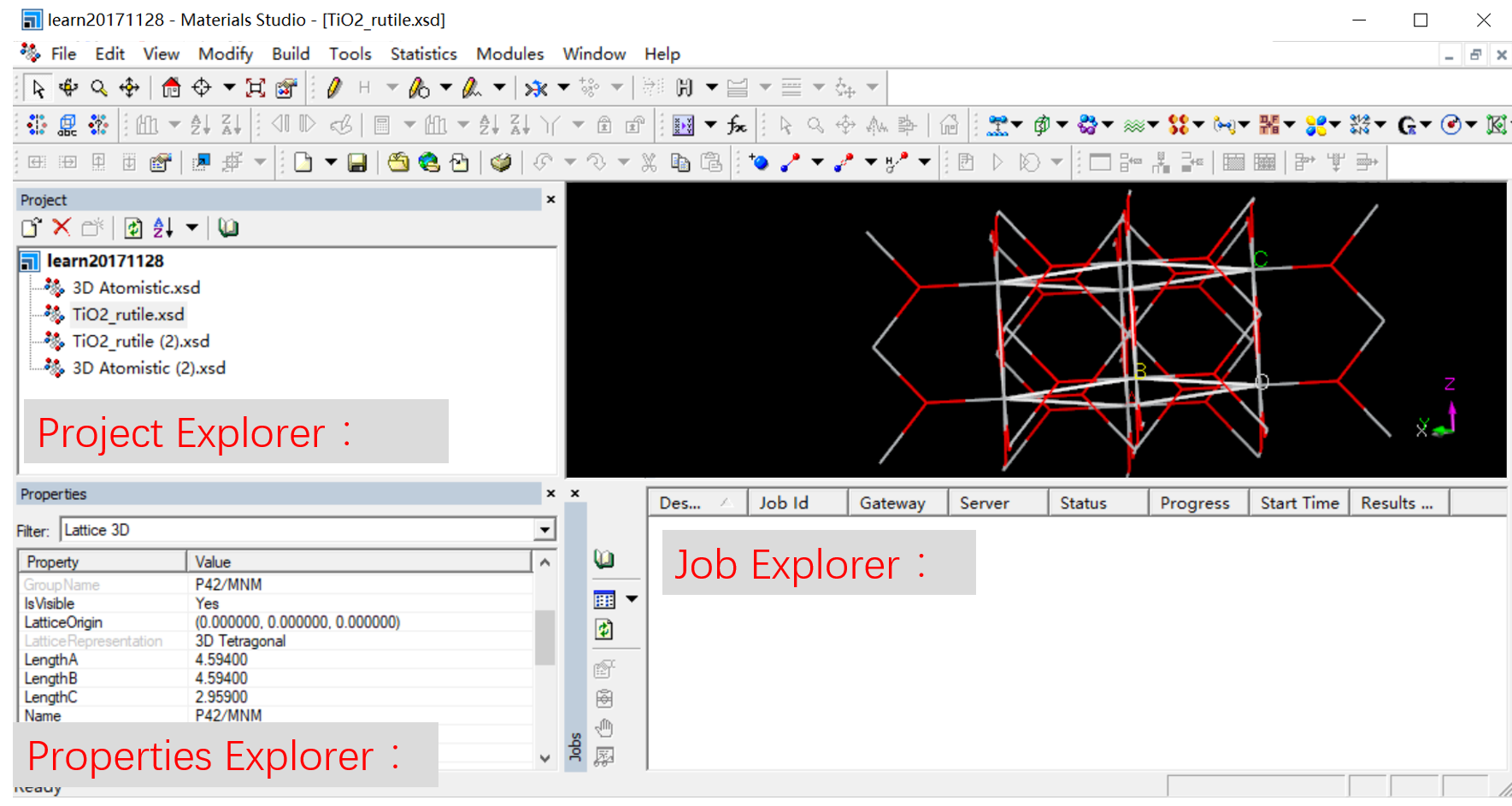
Job Explorer:显示己完成的、正在运行的程序
Project Explorer:显示运行的 job,各种输入与输出文件,近
端远程的状态都可以显示,可以查看结果、修改输出输入的相关设定。
Properties Explorer:材料的原子及电子结构 3D 模型等物性数
据,例如晶体晶胞边长、原子元素种类等等。均可通过双击加以修改
三种窗格如图
20171124第1课
参考
备注
我的集显貌似无法进行图形旋转,建议开启独显
导入TiO2-Rutile
新建项目 File->New Project
保存目录不要有中文
导入TiO2-Rutile,如图,导入C:\Program Files (x86)\Accelrys\Materials Studio 8.0\share\Structures\metal-oxides\TiO2-rutile.msi
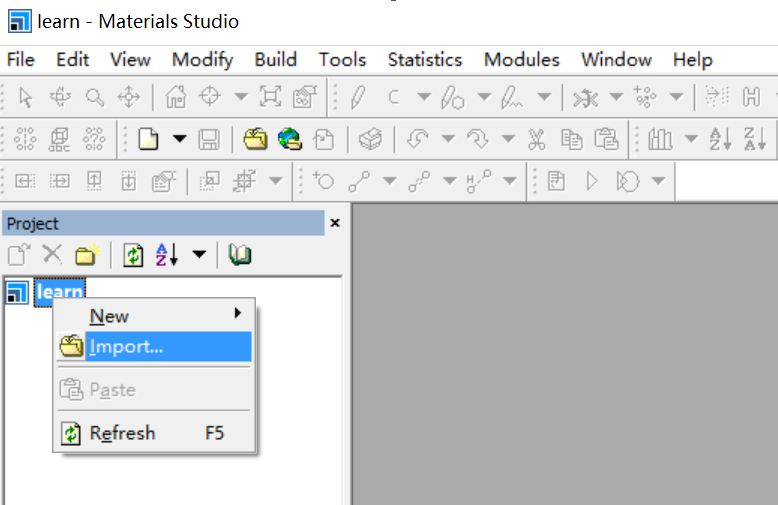
导入后可能看不到原子
在图型上右键->Dispaly Style->Atom->CPK
通过调节CPK的值可以改变原子显示大小
修改
选中原子
左键单击 单选
鼠标拉动选中
shift/ctrl + 单击 多选
如果结构是对称的,修改完也是对称修改
删除原子/制造空位
鼠标单击选中原子,delet键删除
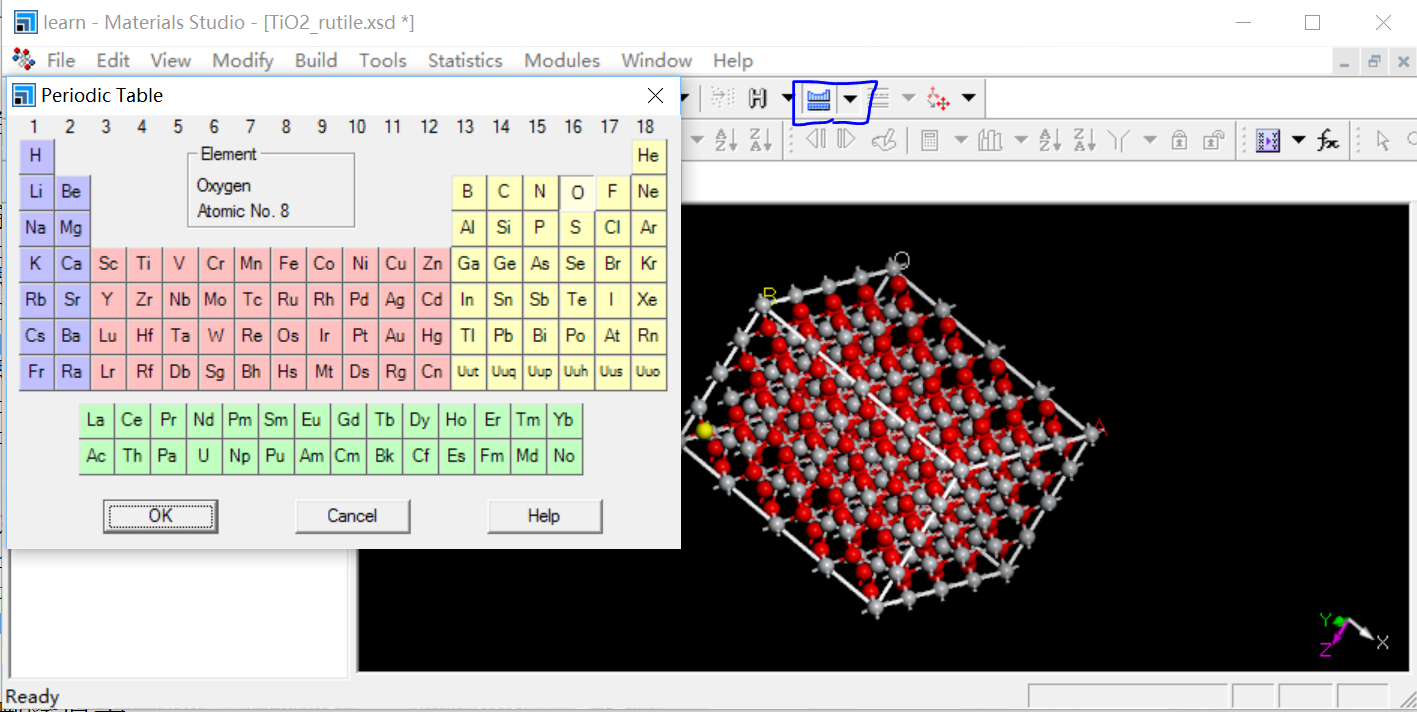
替换原子/掺杂
鼠标单击选中原子,点右上角元素周期表样图标,选中合适元素进行替换,如图
建立超胞
导入TiO2-Rutile后
Build->Symmetry->Supercell调出超胞参数,可以调节ABC三个方向的重复数,例如设为A4B4C4,并适当旋转后如图

TiO2-Rutile表面
1 导入TiO2-Rutile
2 Build->Surfaces->Cleave Surface
主要参数
Cleave(hkl)设为需要的 1 1 0
Top 切割的表面位置(TiO2有三个位置,可以调节此参数发现)
Thinkness 材料厚度(例,6)
3 点击Cleave生成
4 调节合适的参数后Recleave
5 添加真空层
Build->Crystals->Build Vaccum Slab...
主要调节Vaccum thinkness参数
听魏巍老师的解释后我的理解(听听就好,不要信我写的,去请教魏老师):
Vaccum thinkness参数值与第一性原理的一个周期计算单元有关(第一性原理计算必须是周期性结构),数值越大,除去原子后的空白就大,两个周期计算单元之间的原子就不会有干扰如图(实际情况,表面就一层,没有其他层来干扰,而周期性计算会重复出现表面层,空白大可以降低层层间的影响)
另一方面thinkness参数值太大对计算机性能有要求,所以适度吧

6 超胞
上述生成的结构还很小,按照超胞的操作进行拓展,结果如图

20171128第2课
H2O
新建3D Atomistic .xsd
添加O原子
 ,添加后如果找不到了,使用
,添加后如果找不到了,使用
 来调整显示的位置
来调整显示的位置
使用自动加H
 补全H原子,或者使用
补全H原子,或者使用
 进行添加原子
进行添加原子
H2O adsorption on TiO2-Rutile-(110) surface
按照TiO2-Rutile表面的方法生成TiO2的110表面,将H2O步骤建立的水复制到TiO2-Rutile表面的图形中,使用
 移动水分子至其表面
移动水分子至其表面
RuO2-Rutile
导入TiO2-Rutile,使用修改的方法,替换其中一个Ti原子为Ru,由对称性,其他Ti原子会自动被替换
RuO2-TiO2-Rutile (110) interface
建立TiO2-Rutile表面,用鼠标选中右侧所有原子,使用Edite中的方法批量选中部分Ti原子,然后进行替换
如图
20171201第3课
石墨烯
方法一
导入 C:\Program Files (x86)\Accelrys\Materials Studio 8.0\share\Structures\ceramics\graphite.msi石墨结构
Build->Symmtery->Make P1去除结构的对称性
选中一层石墨,删除,就得到了
调整真空层的厚度,即该材料c方向的长度
右键->Lattice Parameter或者Build-Crystals->Rebuild Crystal...,将Parameter选择项卡中的c设置为15或更大值
就得到了单层石墨烯
我觉得再加一点操作:
Build-Symmetry-Find Symmetry...然后选择Find Symmetry可以找回P6/MMM对称性,
方法二
按照之前建立表面的方法,导入后,切出(001)表面,切时厚度设置的薄一点就可以做出单层石墨,然后加上合适厚度的真空层
上课还有一些操作我无法重复出来了,下节课得找老师问问
问题1:
导入石墨->切->加真空层
结果:两个半层石墨,
如何下移一层,然后删掉另一层,保留一层(rebuild)
问题2:
导入石墨->make p1->删除
结果:图片和ppt上不同,如何调整原胞的显示方式
问题3:
这样形成的模型 Lattice 3D中没有P6/MMM对称性,找回对称性又遇到问题1类似的问题
问题解决
移出一层后可通过,Build-Crystals-Rebuild,重新构建
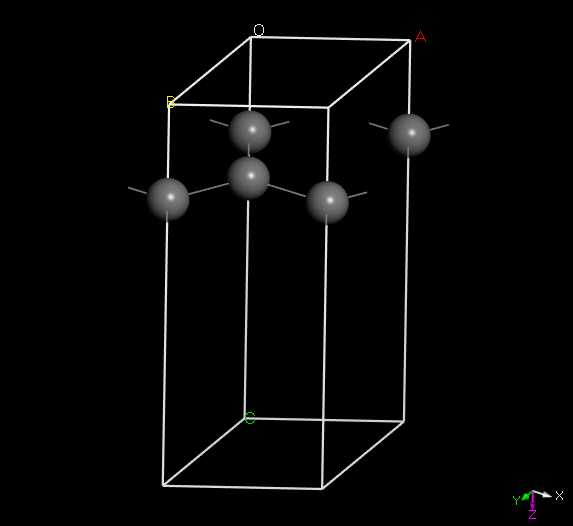
MoS2 (WS2, MoSe2, WSe2…) —TMDs, primitive cell
创建结构
Build-Crystals-Build-Crystal
空间群保持P1默认,Lattice Parameter按照所给参数填入,最后Build
添加原子
使用
 添加原子,选对原子类型和位置,依次添加即可,注意ppt上提供的是分数坐标,Add Atoms 里面Options->Coodinate system选择Fractional分数坐标系
添加原子,选对原子类型和位置,依次添加即可,注意ppt上提供的是分数坐标,Add Atoms 里面Options->Coodinate system选择Fractional分数坐标系
20171205
今天好像没讲什么重要东西
20171208
计算石墨烯的能带
导入
按照导入石墨->make p1->删除一层原子->Find Symmetry得到的石墨烯
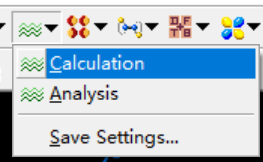
计算
使用CASTEP->Calculation功能如图
参数设置:
Steup->Task->energy
勾选Propeties->Band structure
Properties->More->Path->Creat然后修改下列点为
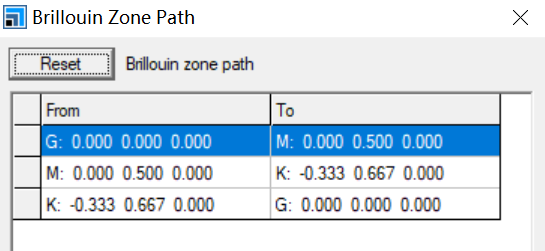
注:该步好像为选择布里渊区,不同的结构该步骤显示的点不同,上图针对使用导入石墨->make p1->删除一层原子->Find Symmetry得到的石墨烯
Run进行计算
查看结果
计算后会生成一个类似xxxx CASTEP Energy的文件夹
选择下面的xxx.xsd文件,然后CASTEP->Analysis选中Band structure再点下面的View查看能带
查看cell文件
上步骤计算得到石墨烯能带结构后,File->Save Project保存计算结果
在计算得到的文件夹上右键Open Containing Folder可以打开计算机中的存储位置
在计算机中进入该文件夹后,查看隐藏文件,里面有个拓展名为cell的文件,里面包含晶格结构等信息
结束
魏老师好像没有继续讲的想法了,感觉上课学的还不如自己看教程来的实在,有点浪费时间
推荐自己搜索教程学习
如果以后自己继续学习DFT相关软件,会在DFT这里更新

















 6875
6875

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









