






作者:顾金杰 中国科学院上海高等研究院(与上海科技大学联合培养)博士研究生
审稿: 阿坤
参考1: https://www. rosettacommons.org/docs /latest/application_documentation/structure_prediction/RosettaCM
参考2: High-resolution comparative modeling with RosettaCM]( http://www. sciencedirect.com/scien ce/article/pii/S0969212613002979 ).
参考Code: http:// FoldTreeHybridize.cc http:// HybridizeProtocol.cc http:// CartesianHybridize.cc
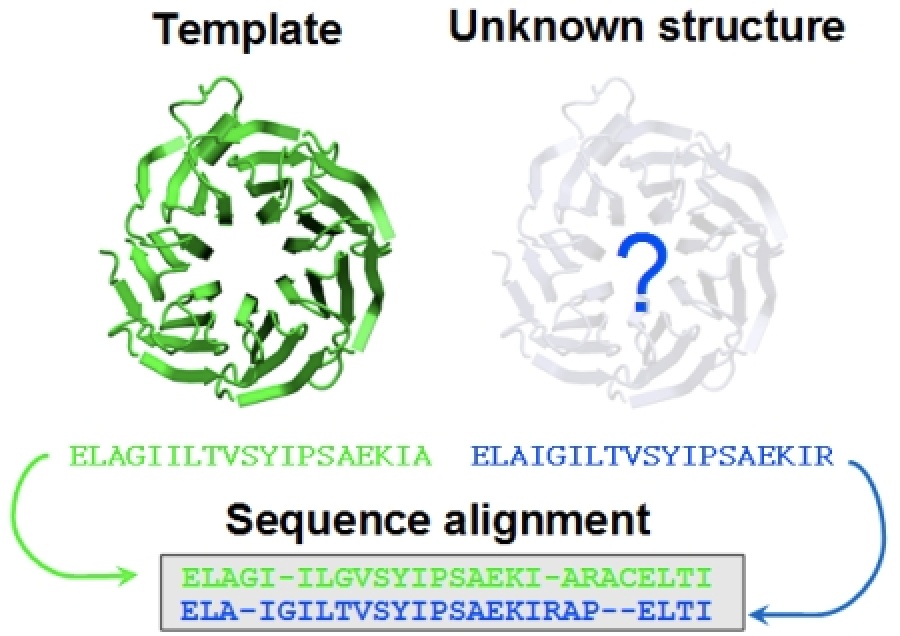
本教程适用于使用单个或多个模板蛋白的结构,预测目的蛋白的同源结构。本文包括两大部分,第一部分是教程教读,第二部分是案例演示。
一、前言与计算原理
1 背景
RosettaCM是Rosetta框架下最新的同源建模方法(2013),其采用了高效的采样方法和全原子力场参数对模型优化,输出高精度的同源模型。RosettaCM在多模板建模中有较大的优势,可以充分利用每个模板的信息。CAP10的实验结果表明,当序列同源性>=15%时,RosettaCM比其他的同源建模方法更优,其模型的骨架和侧链构象更加准确。

2 RosettaCM的计算原理
RosettaCM的核心在于Hybridize, 该算法是一个起始于随机选择的模板,进行蒙特卡洛组装多个模板片段并优化的过程。蒙特卡洛优化过程包括以下几个部分:
- 在非同源部分插入Fragment片段
- 对于随机选择的部分用不同的模板结构进行坐标替代
- 使用平滑的Rosetta矩心能量函数进行笛卡尔空间优化
- 最后,进行所有原子的优化。
RosettaCM计算的详细步骤:
STAGE1
模板的覆盖率是有限的,因此需要将保守的区域提取出来并将缺失的部分补全,形成拥有完整氨基酸序列的结构模型。
- Threading & Alignment: 根据多重序列比对的结果,将目的序列穿线到不同结构模板中,生成碎片式的结构(没有比对上的区域为缺失状态)。然后随机选择(其实可设置每个模板的权重) 一个碎片式结构作为全局比对的目标,将其它模板生成的碎片式结构与初始模型进行叠加,使其重叠度最大化(RMSD最小)。为后续做Segement替换计算三维xyz坐标。
- Make Fragments: 通过FragmentPicker根据序列和二级结构的预测信息,从数据库中生成3和9个长度的短肽片段,后续可用于gap补全、Loop闭环以及Segment骨架优化。
- Make Segments: 从不同碎片式结构中根据二级结构信息对结构进行切割,生成多个二级结构组装单元(Template-derived segments),α-helix至少连续6个氨基酸起算,β-strand至少3个氨基酸起算(不符合标准的片段将被摒弃)。如果二级结构之间存在连接的Loop长度大于3,Loop结构将附着在其附近的α-helix或则β-strand上一起被加载到组装单元片段库中。
- Full length model assembly: 使用Hybridize mover将二级结构组装单元与3/9肽片段进行组装得到序列完整的初始模型,这个过程是通过迭代两种Move完成,第一种是缺失Segment模板的区域使用denovo的Fragment插入(也就是模板无重叠的区域),有共同模板重叠的区域进行二级结构组装单元的坐标替换来采样,通过此方法通常可以获得较为正确的拓扑结构。
STAGE2:
Segment的替换不能保证边界区域和模板拼接模型是符合物理的,因此需要进一步做优化。
- Loop Closure: 随机选取一段骨架区域,根据这段Frame的N端和C端坐标,将denovo fragment或Segments Fragment与其重叠处理,不断地迭代完成闭环。但denovo的fragment重叠是有倾向性的,大概率在当前模型局部键长以及键角能质量较差的区域。
Minimization: 使用平滑的低分辨率能量函数在笛卡尔坐标空间进行能量最小化。
STAGE3:
通过Relax进行全原子模型和能量函数下的骨架微调和侧链Rotamer采样。
二、运行RosettaCM方法
2.1 配置任务
最直接的方法是通过rosetta_tools脚本: protein_tools/scripts/setup_rosettacm.py。
只需要准备以下输入文件:
- 符合格式的序列比对文件
- 模版PDB文件
- 目的蛋白序列文件,fasta格式
Rosetta/tools/protein_tools/scripts/setup_RosettaCM.py [-h] --fasta FASTA
[--alignment ALIGNMENT]
[--alignment_format ALIGNMENT_FORMAT]
[--templates [TEMPLATES [TEMPLATES ...]]]
[--rosetta_bin ROSETTA_BIN] [--build BUILD]
[--platform PLATFORM] [--compiler COMPILER]
[--compiling_mode COMPILING_MODE]
[--setup_script SETUP_SCRIPT] [-j J] [--run]
[--keep_files] [--run_dir RUN_DIR] [--equal_weight]
[--use_dna] [--verbose]注意:
--alignment ALIGNMENT 此处是序列比对文件,作为输入文件,其格式必须符合以下格式中的一种:grishin, modeller, vie, hhsearch, clustalw, fasta.
--templates [TEMPLATES [TEMPLATES ...]]] 此处罗列的是模板结构的PDB文件,比对文件中的模板序列名称必须和模板PDB文件名称一致。
--rosetta_bin ROSETTA_BIN 此处是Rosetta bin目录的路径。
生成完必要的文件后,推荐进行检查有无错误!
2.2 运行RosettaCM
setup_CM脚本运行结束后,切换到rosetta_cm目录,运行以下命令即可,一般来说nstruct需要设置为1000或以上。
Rosetta/main/source/bin/rosetta_scripts.linuxgccrelease @flags -nstruct 1000三 实战案列演示--四聚体蛋白同源建模
选择目的蛋白是来源于某细菌,命名为ABCDE,ABCDE的晶体结构目前还没有被解析。ABCDE氨基酸序列如下:
>ABCDE
MQPTYTIGDYLLDRLVDCGIDRLFGVPGDYNLQFLDRVIAHSALGWVGCANELNAAYAADGYARIKGAGALLTTYGVGELSALNGVAGSYAEHIPVLHIVGAPSTGAQQRGELLHHTLGDGDFRHFARMSEQITCSQALLTAGNACHEIDRVLRDMLTHHRPGYLMLPADVARAAAIAPAQRLLVEAAPADENQLAGFCEHASRLLRGSRRISLLADFLAQRYGLQNTLREWVAKTPVAHATMLMGKGLFDEQQRGFVGTYSGIASAPQTREAIENADTIICIGTRFTDTITAGFTQHLARDKTIEIQPFAVRVGDHWFSGVPMDQALAALMTLSAPLAAEWAAPQVVAPEVEEGADGELTQKNFWATVQGALRPGDIILADQGTAAFGIAALKLPSEASLIVQPLWGSIGFTLPAAYGAQTAAAERRVVLIVGDGAAQLTIQEMGSMLRDKQKPLILLLNNEGYTVERAIHGPEQRYNDIALWDWRRLPEAFAPDVASRCWRVTHTDELREAMAESITSDMLTLVEVMLPKMDIPDFLRAVTQALEERNSRV3.1 模板搜索与比对
在 NCBI 的 protein BLAST 中输入ABCDE的氨基酸序列,并选择 Database 为 Protein Data Bank proteins(pdb)。检索结果如下:

以上这些蛋白是与ABCDE具有高同源性,并且蛋白结构已经被解析(同源建模的时候一般选择的模板同源性需要高于30%,并且尽可能使用更多的模板)。由于在此只是案例演示,我们选择BLAST结果前两个为模板,这两个模板是的PDB ID是1OVM 和2VBF。
选择PROMALS3D multiple sequence and structure alignment server http://prodata.swmed.edu/promals3d/promals3d.php 生成目的蛋白与模板的序列比对文件。
将目的蛋白与模板的序列依次按fasta格式输入到提交框中,运行时间大概在3-5min。
得到fasta格式的序列比对文件。结果如下所示。
>ABCDE
--------------------MQPTYTIGDYLLDRLVDCGIDRLFGVPGDYNLQFLDRVIAHSALGWVGCANELNAAYAADGYARIKGAGALLTTYGVGELSALNGVAGSYAEHIPVLHIVGAPSTGAQQRGELLHHTLGDGDFRHFARMSEQITCSQALLTAGNACHEIDRVLRDMLTHHRPGYLMLPADVARAAAIAPAQRLLVEAAPADENQLAGFCEHASRLLRGSRRISLLADFLAQRYGLQNTLREWVAKTPVAHATMLMGKGLFDEQQRGFVGTYSGIASAPQTREAIENADTIICIGTRFTDTITAGFTQHLARDKTIEIQPFAVRVGDHWFSGVPMDQALAALMTLSAPLAAEWAAPQVVAPEVEEGADGELTQKNFWATVQGALRPGDIILADQGTAAFGIAALKLPSEASLIVQPLWGSIGFTLPAAYGAQTAAAERRVVLIVGDGAAQLTIQEMGSMLRDKQKPLILLLNNEGYTVERAIHGPEQRYNDIALWDWRRLPEAFAP-DVASRCWRVTHTDELREAMAESITS-DMLTLVEVMLPKMDIPDFLRAVTQALEERNSRV
>1OVMA
--------------------MRTPYCVADYLLDRLTDCGADHLFGVPGDYNLQFLDHVIDSPDICWVGCANELNASYAADGYARCKGFAALLTTFGVGELSAMNGIAGSYAEHVPVLHIVGAPGTAAQQRGELLHHTLGDGEFRHFYHMSEPITVAQAVLTEQNACYEIDRVLTTMLRERRPGYLMLPADVAKKAATPPVNALTHKQAHADSACLKAFRDAAENKLAMSKRTALLADFLVLRHGLKHALQKWVKEVPMAHATMLMGKGIFDERQAGFYGTYSGSASTGAVKEAIEGADTVLCVGTRFTDTLTAGFTHQLTPAQTIEVQPHAARVGDVWFTGIPMNQAIETLVELCKQHVHAGLMSSSSGAIPFPQPDGSLTQENFWRTLQTFIRPGDIILADQGTSAFGAIDLRLPADVNFIVQPLWGSIGYTLAAAFGAQTACPNRRVIVLTGDGAAQLTIQELGSMLRDKQHPIILVLNNEGYTVERAIHGAEQRYNDIALWNWTHIPQALSL-DPQSECWRVSEAEQLADVLEKVAHH-ERLSLIEVMLPKADIPPLLGALTKALEACNNA-
>2VBFA
MGSSHHHHHHSSGLVPRGSHMASMYTVGDYLLDRLHELGIEEIFGVPGDYNLQFLDQIISREDMKWIGNANELNASYMADGYARTKKAAAFLTTFGVGELSAINGLAGSYAENLPVVEIVGSPTSKVQNDGKFVHHTLADGDFKHFMKMHEPVTAARTLLTAENATYEIDRVLSQLLKERKPVYINLPVDVAAAKAEKPALS-LEKESSTTNTTEQVILSKIEESLKNAQKPVVIAGHEVISFGLEKTVTQFVSETKLPITTLNFGKSAVDESLPSFLGIYNGKLSEISLKNFVESADFILMLGVKLTDSSTGAFTHHLDENKMISLNIDEGIIFNKVVEDFDFRAVVSSLSELKGIEYEGQYID--KQYEEFIPSSAPLSQDRLWQAVESLTQSNETIVAEQGTSFFGASTIFLKSNSRFIGQPLWGSIGYTFPAALGSQIADKESRHLLFIGDGSLQLTVQELGLSIREKLNPICFIINNDGYTVEREIHGPTQSYNDIPMWNYSKLPETFGATEDRVVSKIVRTENEFVSVMKEAQADVNRMYWIELVLEKEDAPKLLKK------------此处也可以使用别的服务器,如hhpred等。
3.2 生成对称性定义文件(选做)
模板1OVM是同源四聚体,2VBF是同源二聚体。根据文献,目的蛋白ABCDE也是四聚体,考虑到目的蛋白与1OVM的同源性高达62.12%,因此选择1OVM四聚体结构生成对称性定义文件(symmetry definition file) 。
运行命令:
perl $ROSETTA/main/source/src/apps/public/symmetry/make_symmdef_file.pl -m NCS -p 1ovm.pdb -a A -i B C D > 1ovm.symm其中,1ovm.pdb为所选择用于产生对称性定义文件的模板结构文件。运行此命令后得到文件名为1ovm.symm的对称性定义文件。
3.3 建立RosettaCM任务
此案例选择运行setup_CM.py生成threaded模型。运行setup_CM.py时输入的模板的结构和序列皆为单链结构,在运行Hybridize mover时将对称性定义文件添加至XML文件,则会得到多聚体的模型。
特别注意: setup_CM.py识别的文件名需要大于5个字符,当文件名不足5个字符时,往往会报错,因此将1OVM的单链PDB命名为1OVMA,将2VBF单链PDB命名为2VBFA)
运行setup_CM.py
输入文件为:
- ABCDE.fasta #目的蛋白序列文件
- ABCDE_alignment.fasta #目的蛋白与模版序列比对文件
- 1ovmA.pdb #模板1结构文件(A链)
- 2vbfA.pdb #模板2结构文件(A链)
运行命令为:
python2 $ROSETTA/tools/protein_tools/scripts/setup_RosettaCM.py --fasta ABCDE.fasta --alignment ABCDE_alignment.fasta --alignment_format fasta --templates 1OVMA.pdb 2VBFA.pdb --rosetta_bin $ROSETTA/main/source/bin --platform macos --compiler clang --build mpi输出一个名为rosetta_cm的文件夹,该文件夹中含有以下文件:
- 1OVMM_thread.pdb #模板1线状模型
- 2VBFM_thread.pdb #模板2线状模型
- converted_alignment.aln #aln格式的序列比对文件
- flags #flag文件
- rosetta_cm.xml #XML文件
将1ovm.symm文件移入rosetta_cm文件夹中。
将XML文件中的symmetric="0"改为symmetric="1"
<SCOREFXNS>
<ScoreFunction name="stage1" weights="score3" >
<Reweight scoretype="atom_pair_constraint" weight="0.1"/>
</ScoreFunction>
<ScoreFunction name="stage2" weights="score4_smooth_cart">
<Reweight scoretype="atom_pair_constraint" weight="0.1"/>
</ScoreFunction>
<ScoreFunction name="fullatom" weights="beta_cart" >
<Reweight scoretype="atom_pair_constraint" weight="0.1"/>
</ScoreFunction>
</SCOREFXNS>即为:
<SCOREFXNS>
<ScoreFunction name="stage1" weights="score3" symmetric="1">
<Reweight scoretype="atom_pair_constraint" weight="0.1"/>
</ScoreFunction>
<ScoreFunction name="stage2" weights="score4_smooth_cart" symmetric="1">
<Reweight scoretype="atom_pair_constraint" weight="0.1"/>
</ScoreFunction>
<ScoreFunction name="fullatom" weights="beta_cart" symmetric="1">
<Reweight scoretype="atom_pair_constraint" weight="0.1"/>
</ScoreFunction>
</SCOREFXNS>将Template部分的加上symmdef="1ovm.symm"。
<Template pdb="/Users/kunkun/Desktop/rosettaCM/rosetta_cm/1OVMA_thread.pdb" cst_file="AUTO" weight="1.0" />
<Template pdb="/Users/kunkun/Desktop/rosettaCM/rosetta_cm/2VBFA_thread.pdb" cst_file="AUTO" weight="1.0" />改为:
<Template pdb="/Users/kunkun/Desktop/rosettaCM/rosetta_cm/1OVMA_thread.pdb" cst_file="AUTO" weight="1.0" symmdef="1ovm.symm"/>
<Template pdb="/Users/kunkun/Desktop/rosettaCM/rosetta_cm/2VBFA_thread.pdb" cst_file="AUTO" weight="1.0" symmdef="1ovm.symm"/>此外,如果得知某个结构的可信度更好,可以将其weight调高,增加作为初始模板的概率,此操作可以提高计预测的精度。
将路径切换至rosetta_cm文件夹,运行命令:
rosetta_scripts.mpi.macosclangrelease @flags -nstruct 2由于是案例演示,-nstruct 输出结果数设置为2,实际工作中,通常需要输出大量结构进行比较选择推荐1000以上。模型的选择主要根据打分进行排名,将前10%的结构进行聚类计算,将每个最大簇的中心构象取出,作为最佳输出的模型候选。
得到目的蛋白的模型结构后,可以选择SAVES https://servicesn.mbi.ucla.edu/SAVES 等工具对其进行评价。














 2592
2592











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









