







了解 AmberTools 从何处开始,这主要是管理软件包中信息传递的问题,请参见图 1.1。首先需要了解模拟程序(sander、pmemd、mdgx 或 nab)需要哪些信息。 您需要知道这些信息从何而来,又是如何以这些程序所需的形式出现的。 本节旨在为新用户提供指导,不能替代各个程序的文档。
所有模拟程序都需要的信息(见图 1.1 中的圆圈):
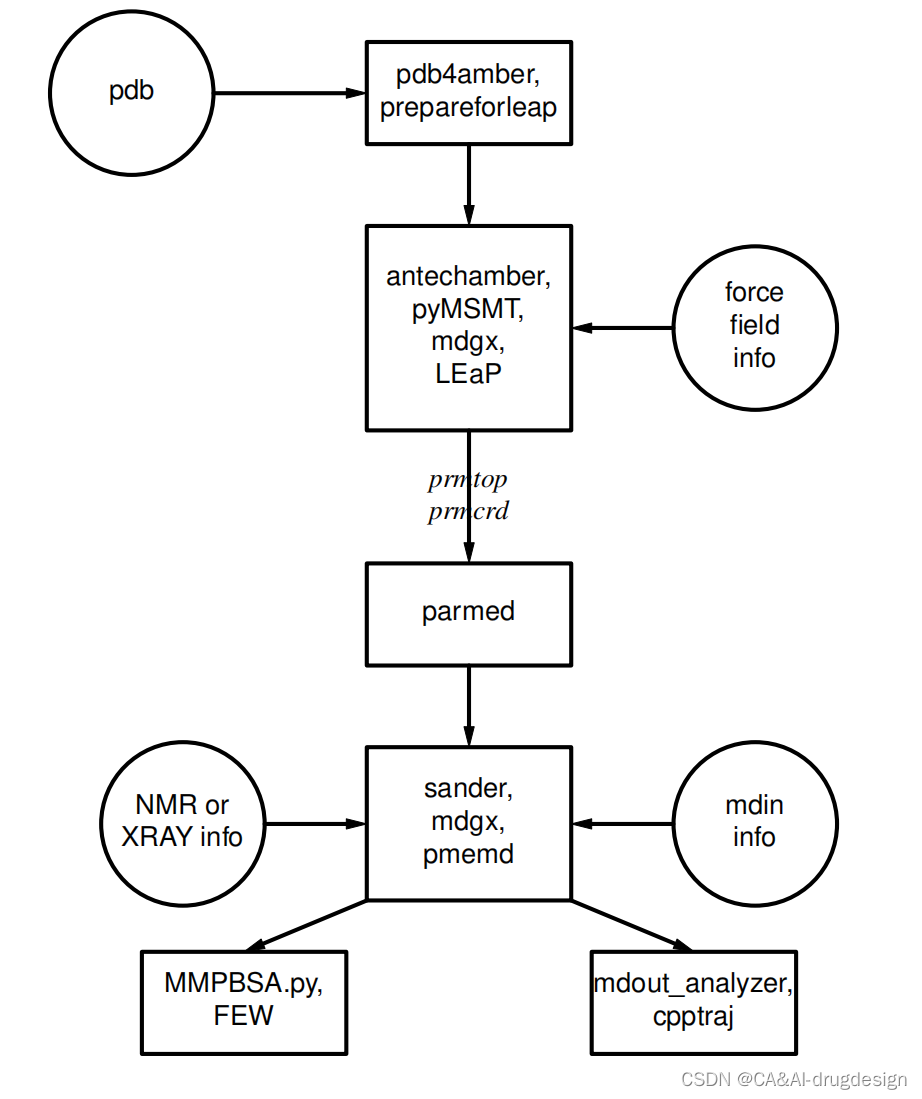
图1.1 Amber的基本信息传递
-
系统中每个原子的笛卡尔坐标。 这些坐标通常来自 X 射线晶体学、核磁共振光谱或模型建立。这些坐标一般应为 Protein DataBank(PDB)格式。LEaP 程序提供了执行许多建模任务的平台,但用户也可以考虑使用其他程序。 一般来说,需要对这些文件进行编辑,pdb4amber 脚本可以完成部分编辑工作。
-
拓扑结构: 连接性、原子名称、原子类型、残基名称和电荷。这些信息来自 $AMBERHOME/dat/leap/lib 目录下的数据库,详见第 3 章。 该数据库包含标准氨基酸、N 端和 C 端带电氨基酸、DNA、RNA 以及常见糖类和脂类的拓扑信息。其他分子的拓扑信息(标准数据库中没有)保存在用户生成的 "residue files "中,这些文件通常使用 antechamber 创建。
-
力场: 力场:系统中所有键、角、二面体和原子类型的参数。 多个力场的标准参数可在 $AMBERHOME/dat/leap/parm 目录中找到;更多信息请参见第 3 章。 这些文件可 "按原样 "用于蛋白质和核酸,用户也可自行准备包含对标准力场修改的文件。
-
**拓扑和坐标文件(通常称为 prmtop 和 prmcrd,但也可使用任何合法文件名)**创建完成后,可使用 parmed 脚本检查和验证这些文件并进行修改。尤其是检查有效性(checkValidity)操作会标记出许多潜在的问题。
-
命令: 用户指定所需的程序选项和状态参数。 这些参数在输入文件(默认名为 mdin)或用 NAB 语言编写的 "driver "程序中指定。















 338
338











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









